
Инженерный тур. 2 этап
Пандемия коронавирусной инфекции стала трудным испытанием для системы здравоохранения. Компании рынка «Хелснет» НТИ внесли значительный вклад в решение задач по ПЦР- и ИФА-диагностике коронавирусной инфекции, разработке новых вакцин. Большую роль в этом сыграли не только специалисты, работающие в молекулярно-биологических лабораториях, но и биоинформатики.
Сквозная технология Национальной технологической инициативы (НТИ) «Управление свойствами биологических объектов» основана на достижениях инженерной биологии: генетической инженерии, биотехнологии, молекулярной биологии, биохимии и многих других. Актуальность исследований в области разработки новых инструментов генетической инженерии и геномного редактирования сегодня не вызывает сомнений. Большая часть новых продуктов фармацевтической индустрии, биотехнологий и других отраслей основана на использовании инструментов, созданных в конце XX века (генетическая инженерия), сегодня активно разрабатывают и другие подходы, среди которых особое место занимает синтетическая биология и технология геномного редактирования.
Командные задачи первого блока для 8–9 классов открыты для решения. Соревнование доступно на платформе Яндекс.Контест: https://contest.yandex.ru/contest/69915/enter/.
В живой клетке одновременно происходят различные биохимические процессы — от дыхания до пищеварения.

Какие процессы в клетке относят к матричным?
Выберите верные варианты ответа:
- Фотосинтез.
- Репликация ДНК.
- Обратная транскрипция.
- Кроссинговер.
- Сплайсинг.
B, C.
В 1959 году Артуру Корнбергу была присуждена Нобелевская премия по физиологии и медицине «за открытие механизма биологического синтеза рибонуклеиновой и дезоксирибонуклеиновой кислот». Работая с командой в Вашингтоне, Корнберг искал ферменты, которые собирают нуклеотиды в РНК или ДНК. Спустя некоторое время ученому удалось успешно построить ДНК по матрице ДНК при помощи ДНК-полимеразы и описать ее.

Какую ошибку совершил А. Корнберг, описавший ДНК-полимеразу?
Выберите верные варианты ответа:
- Артур Корнберг открыл лишь одну ДНК-полимеразу из трех (пяти), которая есть у E. coli.
- Открытая ДНК-полимераза I не работает в качестве репликативной.
- Артур Корнберг не учел, что для активности ДНК-полимеразе требуется \(\ce{Mg^2+}\).
- Субстрат, используемый Артуром Корнбергом, не является физиологическим.
- Репликация происходит полунепрерывно, а не непрерывно, как в модели А. Корнберга.
A, B.
Долгое время ученые, изучавшие репликацию ДНК, не могли понять, каким образом обе цепи ДНК могут реплицироваться одновременно. Ведь они антипараллельны, а ДНК-полимераза может присоединять нуклеотиды только к 3\(^\prime\)-концу, значит, она может удлинять только одну из двух растущих цепей!
На этот вопрос смог ответить Рейджи Оказаки, который провел ряд экспериментов и выяснил, что при репликации у бактерий большая часть новообразованной ДНК обнаруживается в форме небольших кусков, названных фрагментами Оказаки.
Какие суждения верны для фрагментов Оказаки?
Выберите верные варианты ответа:
- Фрагменты Оказаки эукариот длиннее, чем у прокариот.
- Фрагменты Оказаки эукариот короче, чем у прокариот.
- Длина фрагмента Оказаки постоянна у всех живых организмов.
- Длина фрагмента Оказаки зависит от скорости синтеза ДНК-полимеразой.
- Фрагменты Оказаки в отстающей цепи длиннее, чем в лидирующей цепи.
B, D.
В лаборатории для многих целей используются центрифуги или микроспины (microspin). Принцип их действия основан на центробежной силе, что позволяет осаждать раствор в пробирке или разделять компоненты раствора по массе (например, клетки от среды для культивирования). Для того чтобы эти приборы работали исправно, при расстановке пробирок нужно следить за тем, чтобы они были в равновесии.
На рис. 1.4 изображен полностью загруженный микроспин на 12 положений.

Необходимо разместить в этом приборе три пробирки одинаковой массы таким образом, чтобы он находился в равновесии. Выберите из предложенных вариантов номера положений, в которых можно разместить пробирки без угрозы. Для удобства будем вести отсчет положений от помеченного стрелкой и по часовой стрелке.
- 1, 5, 9.
- 1, 7, 10.
- 4, 12, 8.
- 9, 7, 11.
- 2, 8, 5.
Необходимо из предложенных вариантов выбрать те, где цифрами обозначены положения, которые являются вершинами правильного треугольника. Это нужно для сохранения равновесия прибора.
A, C.
Одним из самых популярных и используемых методов секвенирования ДНК долгое время был метод Сэнгера, за который в 1980 году была присуждена Нобелевская премия. Этот метод позволяет считывать последовательности до 1000 пар нуклеотидов и потому активно используется для секвенирования и по сей день, например, отдельных участков генома для анализа мутаций.
При проведении секвенирования по Сэнгеру реакционную смесь разделяют методом капиллярного гель-электрофореза. На рис. 1.5 приведена «секвенирующая лестница», по которой можно определить последовательность ДНК.

Определите по результату секвенирования на рис. 1.5 последовательность цепи. В ответ запишите последовательность, комплементарную данной в направлении \(3^\prime\rightarrow5^\prime\).
Идя снизу вверх по рисунку, определить по бэндам последовательность ДНК.
Список литературы
- https://biomolecula.ru/articles/metody-v-kartinkakh-sekvenirovanie-nukleinovykh-kislot (pdf-версия веб-страницы https://disk.yandex.ru/i/-27yN3oOeu5aFw).
TAACGAATGCATGCAATCAGCATGT.
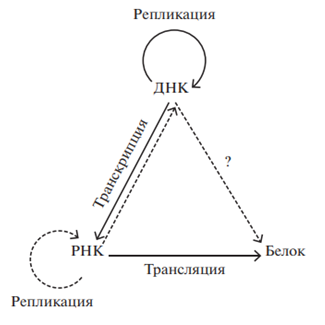
В 1958 году Фрэнсис Крик выдвинул правило, которое к 1970-му году стало центральной догмой молекулярной биологии.
Переход генетической информации последовательно от ДНК к РНК и затем от РНК к белку является универсальным для всех без исключения клеточных организмов, лежит в основе биосинтеза макромолекул. Репликации генома соответствует информационный переход ДНК \(\rightarrow\) ДНК или РНК \(\rightarrow\) РНК.
Как известно, гены кодируются в направлении \(5^\prime\rightarrow3^\prime\) нуклеотидной последовательности и ограничиваются старт- и стоп-кодонами по краям. Открытая рамка считывания (англ. Open Reading Frame, ORF) представляет собой последовательность нуклеотидов в составе ДНК или РНК, потенциально способную кодировать белок. ORF начинается старт-кодоном — самым первым триплетом, с которого запускается трансляция, и заканчивается последним триплетом перед стоп-кодоном, который не входит в состав ORF.
В представленной ниже нуклеотидной последовательности найдите возможные варианты ORF и транслируйте их, используя правила генетического кода.
5\(^\prime\)-TAAACTCCGATATTACATCAGTACATACTCCCCAGCCCCCACGCAAACCCGGAGGTACCTGAAGCCAGCAGCCATAATTGCCGAAGCTACTTCTATCCGTTGGGGGAACGGCCGTAGCTACTGCGATGCACGAGATTTTATAAATGCATTATGATCGAGCATAAATGATATAAATGGCGATGTAAATGTGGATCAATAACGAGCGCTAA-3\(^\prime\)
В ответе последовательно приведете аминокислотные последовательности в верхнем регистре, разделив их дефисом и опустив старт-кодоны. Открытыми рамками считывания на обратной цепи следует пренебречь.
Пример ответа: TNW-AGPC-FlAM.
Используя программу UGENE, найти все ORF и с помощью таблицы генетического кода определить аминокислотную последовательность.
Список литературы
- Генетический код — статья из Википедии https://ru.wikipedia.org/wiki/Генетический_код.
- Открытая рамка считывания — статья из Википедии https://ru.wikipedia.org/wiki/Открытая_рамка_считывания.
- Предлагаемый софт для решения задачи — UGENE http://ugene.net/ru/.
HEIL-HYDRA-I-AM-A-WINNER.
Белок Rad51, продукт гена RAD51 человека (https://www.ncbi.nlm.nih.gov/gene/5888), принимает участие в репарации двуцепочечных разрывов в ДНК.
Изучите информацию о данном белке в базе данных NCBI, определите, на какой хромосоме человека находится этот ген. Введите номер хромосомы.
15.
Белок Rad51, продукт гена RAD51 человека, принимает участие в репарации двуцепочечных разрывов в ДНК по пути гомологичной рекомбинации. Повышенный уровень экспрессии данного гена увеличивает эффективность геномного редактирования.
Изучите информацию о мРНК данного гена в базе данных NCBI (https://www.ncbi.nlm.nih.gov/nuccore/NM_001164269.2), определите, сколько остатков аминокислот кодирует основная открытая рамка считывания (CDS). Введите число остатков аминокислот.
340.
Командные задачи второго блока для 8–9 классов открыты для решения. Соревнование доступно на платформе Яндекс.Контест: https://contest.yandex.ru/contest/70124/enter/.
Метод секвенирования, придуманный Сэнгером, не сразу был удобен для использования. Первоначально был разработан «плюс-минус» метод секвенирования, который состоял из двух этапов: на первом этапе проводили четыре реакции ПЦР в присутствии разных дезоксинуклеозидтрифосфатов с радиоактивной меткой и четыре реакции ПЦР в отсутствии каждого из них, после чего проводили очистку.

Для сокращения числа манипуляций и оптимизации процесса через несколько лет Сэнгер предложил другой способ — метод «терминаторов». Он изменил реакционную смесь так, что вместо восьми реакций необходимо было всего четыре.
Что содержится в реакционной смеси для каждой из четырех реакций для метода «терминаторов»?
- ДНК-полимераза.
- Смесь дидезоксинуклеозидтрифосфатов (ddNTP).
- Один из дидезоксинуклеозидтрифосфатов (ddATP, ddTTP, ddCTP или ddGTP).
- Праймеры.
- Смесь меченых 32P дезоксинуклеотидов (dNTPs).
- Один меченый 32P по \(\alpha\)-положению дезоксинуклеотид (dATP, dTTP, dCTP или dGTP).
- Смесь дезоксинуклеотидов (dNTPs).
- ДНК-матрица.
A, C, D, F, G, H.
Рестрикционный анализ плазмид позволяет определить различные параметры плазмиды — ее истинную длину, наличие вставки или мутации и т. д. Рестрикционный анализ — метод определения сайтов узнавания эндонуклеаз рестрикции в ДНК.
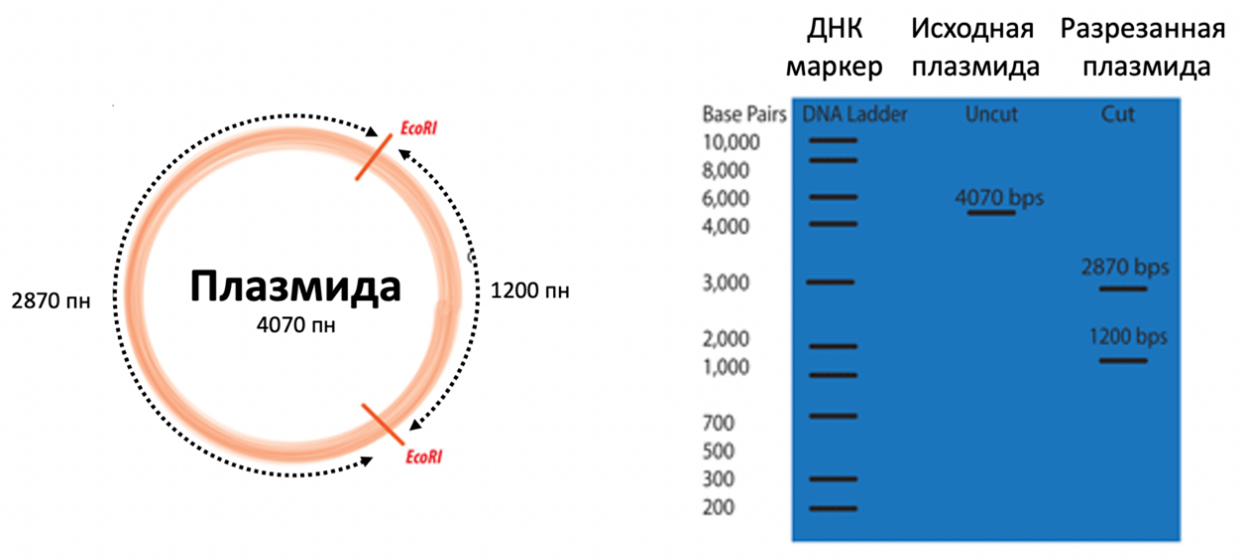
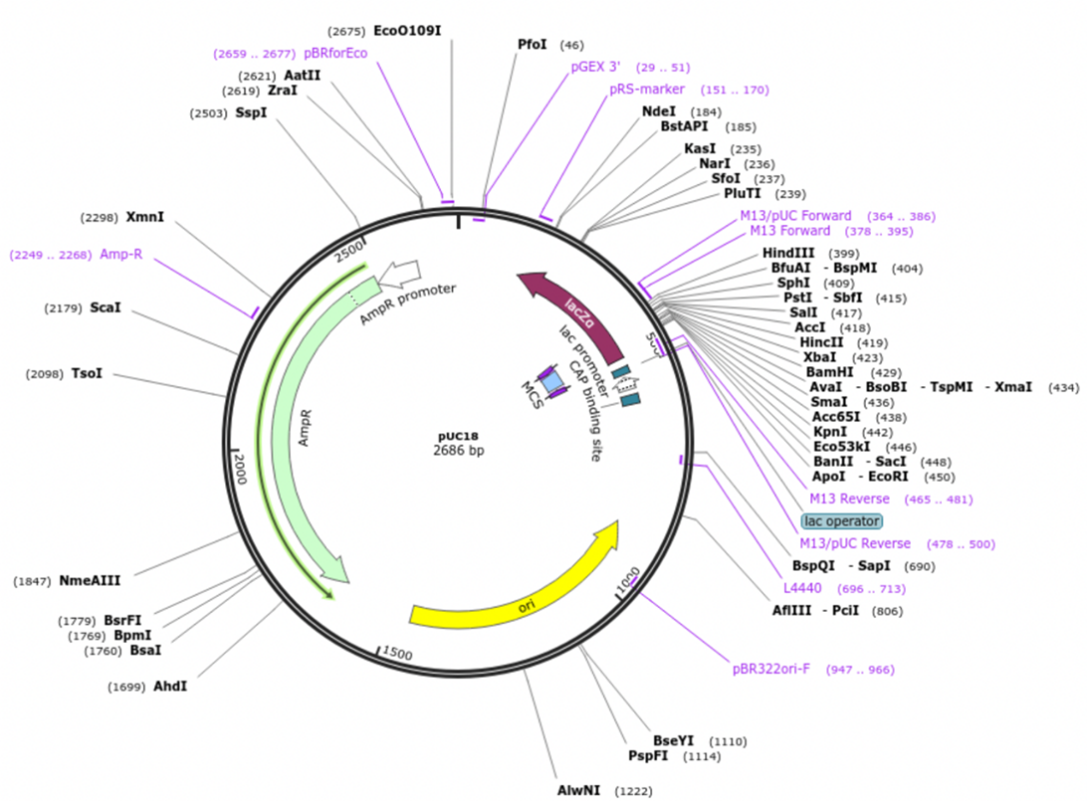
Проведите реакцию рестрикции in silico для плазмиды pUC18 эндонуклеазой рестрикции Rsa I. В ответе укажите длину наименьшего продукта рестрикции без указания размерности.
Пример ответа: 1500.
В предложенной программе UGENE провести рестрикционный анализ с помощью специальных функций программы. Выбрать наименьший по количеству пар нуклеотидов фрагмент.
Список литературы
- Карта плазмиды pUC18 — https://www.addgene.org/50004/ (pdf-версия https://disk.yandex.ru/i/zWWHhcmPhHRs-A).
- Описание эндонуклеазы рестрикции Rsa I — https://sibenzyme.com/product/rsa-i/ (pdf-версия https://disk.yandex.ru/i/V0uI3kJ1Zl4WyQ).
- Предлагаемый софт для решения задачи — UGENE http://ugene.net/ru/.
271.
Определите молекулярную массу полипептида, закодированного следующей нуклеотидной последовательностью: 5\(^\prime\)-AAGATTACCACTTAT-3\(^\prime\).
В ответе укажите число с точностью до одного знака после запятой без указания размерности.
Пример ответа: 110,7 (убедитесь, что у вас именно запятая, а не точка).
С помощью таблицы генетического кода определите последовательность аминокислот в полипептиде, после чего из данных в таблице, которая доступна по ссылке из списка литературы, сложите молекулярные массы аминокислот.
Список литературы
- Молекулярные массы аминокислот — https://ru.wikipedia.org/wiki/Аминокислоты.
696,8.
Плазмида pNTO имеет длину 2024 пн. Эндонуклеаза рестрикции ApoI распознает сайт RAATTY. Предположим, что нуклеотиды в плазмиде расположены случайно и количество A = T = C = G. Определите, сколько сайтов рестрикции ApoI может находиться в плазмиде pNTO. В ответе укажите предполагаемое количество сайтов рестрикции (округлите до ближайшего целого числа).
Пример ответа: 10.
Сайт рестрикции из 6 нуклеотидов RAATTY появляется в одном случае из: \[2\cdot 4\cdot 4\cdot 4\cdot 4\cdot 2 = 1024.\] Вероятность встречи в плазмиде длиной 2024 = \(2024\,/\,1024 = 2\).
2
.
Цикл трикарбоновых кислот, или цикл Кребса — это ключевой этап клеточного дыхания, где происходит окисление питательных веществ для получения энергии.
В начале цикла трикарбоновых кислот ацетил-КоА, образованный при распаде глюкозы, жирных кислот или аминокислот, отдает свою ацетильную группу четырехуглеродному соединению — оксалоацетату (щавелевоуксусной кислоте — ЩУКа), при этом образуется шестиуглеродный цитрат. Цитрат затем изомеризуется в изоцитрат, который далее дегидрируется и декарбоксилируется до пятиуглеродной кислоты — \(\alpha\)-кетоглутарата. \(\alpha\)-кетоглутарат вновь декарбоксилируется, превращаясь в четырехуглеродный сукцинат. Сукцинат затем превращается в фумарат под воздействием сукцинатдегидрогиназы, после чего он в два этапа ферментативно преобразуется снова в оксалоацетат — ЩУКу.
Сукцинатдегидрогеназа является центральным звеном между энергетическим обменом (цикл Кребса) и дыхательной цепью, обеспечивая эффективное использование выделенной энергии для синтеза АТФ. SDH состоит из четырех субъединиц: SDHA, SDHB, SDHC и SDHD. Субъединица А содержит ФАД и сайт связывания сукцината, что крайне важно для ферментативного преобразования сукцината в фумарат.
Всем известно, что цикл трикарбоновых кислот представляет собой один из основных энергетических путей в клетках не только животных, но и растений, и грибов, и бактерий. В связи с этим, проанализируем генетические особенности субъединицы А сукцинатдегидрогеназы (SDH) у ЛЕБЕДЯ-шипуна (Cygnus olor).
Используя базу данных NCBI, определите, на какой хромосоме расположен ген SDHA у ЛЕБЕДЯ-шипуна и количество экзонов в последовательности гена SDHA в аминокислотах. Ответ запишите в формате: [№ хромосомы]_[количество экзонов].
Пример ответа: 10_2.
Ген находится в 2-й хромосоме, содержит 15 экзонов.
2_15.
Когда механизмы регуляции таких путей, как цикл трикарбоновых кислот, нарушаются, то результатом может стать развитие серьезного заболевания. Ферменты цикла кодируются генами домашнего хозяйства, и отсутствие функциональных копий этих генов может быть фатальным. Мутации генов сукцинатдегидрогеназы (SDH) вызывают РАК надпочечников (феохромоцитому). В культурах клеток с такими мутациями накапливается сукцинат, что может привести к формированию нейроэндокринных опухолей, таких как параганглиома, карцинома почек и стромальная опухоль желудочно-кишечного тракта.
Предложена последовательность области гена SDHA из клеток параганглиомы, полученной от лебедя-шипуна (Cygnus olor). Последовательности получали методом ПЦР с пары праймеров и секвенировали методом Сэнгера.
Последовательность (\(5^\prime\rightarrow3^\prime\)) мутантного варианта гена SDHA из Cygnus olor:
>SDHA_mut
CCTGATGCTTTGTGCTCTACAAACAATCTATGGTGCTGAGGCTCGCAAGGAGTCCCGGGG TGCTTATGCCAGAGAGGACTATAAGGTACGGA
Определите последовательности прямого и обратного праймеров, если известно, что праймеры имели длину, равную 20 нт.
В ответ запишите последовательности праймеров (сначала прямого, потом обратного) в направлении \(5^\prime\rightarrow3^\prime\), разделенные запятой без пробелов.
Пример ответа: ATGC,CATG.
Достроить к последовательности гена комплементарную, на прямой цепи выбрать последовательность, на которую будет отжигаться прямой праймер, на комплементарной цепи выбрать место для обратного. Записать в ответ их последовательности.
CCTGATGCTTTGTGCTCTAC,TCCGTACCTTATAGTCCTCT.
Методом сравнения продуктов ПЦР мутантного и дикого вариантов гена SDHA определите мутацию, которая может приводить к образованию параганглиомы у лебедя-шипуна, используя данные и результаты предыдущей части задания.
Ответ введите в формате c.XY>Z, где X — позиция нуклеотида, Y — истинный нуклеотид, Z — мутантный нуклеотид.
Список литературы
- Скачивание последовательности дикого типа: перейти по ссылке https://www.ncbi.nlm.nih.gov/nuccore/NC_049170.1?from=92788988&to=92803671&report=genbank&strand=true \(\Rightarrow\) send to \(\Rightarrow\) coding sequence \(\Rightarrow\) create file.
- Предлагаемый софт для решения задачи: UGENE http://ugene.net/ru/.
c.1765T>C.
Трансформация бактерий — это процесс, при котором бактерия принимает чужую ДНК из окружающей среды в свой генетический аппарат. Это позволяет бактериям приобретать новые свойства такие, как устойчивость к антибиотикам или дополнительные метаболические возможности.
В лабораторных условиях искусственная трансформация дает возможность вводить новые гены, изучать функции генома и создавать бактерии с полезными свойствами для биотехнологий. Существует два основных метода искусственной трансформации.
Первый метод — химически-тепловая трансформация, где с помощью химических веществ и теплового воздействия вводят чужеродную ДНК в клетку. Второй метод — электропорация, при которой электрический импульс временно делает мембрану клетки проницаемой, позволяя ДНК проникать внутрь.
Эффективность трансформации (ЭТ) оценивается по количеству трансформированных клеток, выраженному в колониеобразующих единицах (КОЕ) на микрограмм введенной плазмидной ДНК. \[\text{ЭТ}= \frac{\text{КОЕ}}{\text{количество трансформируемой плазмиды в мкг}}.\]
После приготовления химически компетентных клеток E. coli штамма BL21 (DE3) проверяли эффективность их трансформации при помощи введения стандартного плазмидного вектора pUC19 (2686 пн). Для этого приготовили 100-кратное разведение плазмиды со стоковой концентрацией 2024 нг/мкл. К химически компетентным клеткам добавили 2,5 мкл разведенной плазмиды, после чего 1/20 от общего количества трансформированных клеток посеяли на чашки Петри. Спустя 16 ч инкубации чашек Петри при 37 °С провели подсчет колоний, который составил 347 КОЕ.
Определите эффективность химической трансформации. Ответ округлите до целого числа.
137154.
Лаборант Василий мечтал стать ученым. После поступления в университет ему не терпелось начать работать и, как только представилась возможность, устроился в лабораторию профессора Живодерова. Живодеров был особенным научным руководителем и любил ставить новичков на непростые задачи. Новоиспеченному лаборанту пришлось иметь дело с Аполипопротеином C1 (ApoC1). Это белок, входящий в состав липопротеинов плазмы крови, участвует в транспорте липидов через кровь, а также в регулировании активности липопротеин-связанных ферментов. Василий преобразовывал его в цепь аминокислот и начинал обрабатывать ферментами. Сначала он обработал химотрипсином, потом из полученных цепей взял самую длинную и обработал ее глутамил-эндопептидазой (Глу-C (эндопептидаза V8). Потом обработал получившиеся полипептиды флуорексоном, провел детекцию и выбрал тот, у которого первой аминокислотой был Gly. Этот полипептид он хотел обработать карбоксипептидазой, но отвлекся на птичку в окне и случайно сделал это дважды.
Задача. Повторите эксперимент Василия. Найдите аминокислотную последовательность белка в NCBI: изоформа NP_001307995.1. Помните, что химотрипсин гидролизует после остатков ароматических аминокислот: фенилаланина (Phe), тирозина (Tyr) и триптофана (Trp); глутамил-эндопептидаза гидролизует после глутаминовой кислоты (Glu) и аспарагиновой кислоты (Asp); флуорексон позволяет «узнать» аминокислоту с N-конца, карбоксипептидаза отрезает аминокислоту с C-конца.
Какую аминокислоту Василий «потерял» из-за невнимательности?
Выберите верный вариант ответа:
- Asp.
- Val.
- Ser.
- Pro.
- Lys.
- Glu.
- Leu.
- Trp.
- Asn.
- Ile.
- Ala.
- Gln.
D.
Повторы в геноме — это последовательности ДНК, которые встречаются многократно, часто подряд или в разных частях генома. У человека около 50% генома состоит из повторяющихся элементов, включая простые тандемные повторы и сложные мобильные элементы такие, как транспозоны. У других видов этот процент может варьироваться: у некоторых растений и амфибий повторы могут занимать более 80% генома, тогда как у бактерий их доля минимальна.
Повторы играют важную роль в эволюции генома, способствуя его структурным изменениям, генетической рекомбинации и регуляции генов. Некоторые из них, такие как тандемные повторы, используются в судебной экспертизе и популяционной генетике, а другие, например, LINE- и SINE-элементы, влияют на экспрессию генов. Хотя повторы иногда вызывают мутации и генетические заболевания, они также обеспечивают гибкость генома и участвуют в его адаптации.
Маскирование повторов в геноме необходимо для корректного анализа уникальных участков ДНК, так как повторы могут вызывать ошибки при сборке генома, картировании и анализе данных секвенирования. Например, при выравнивании ридов на референсный геном повторы могут привести к множественным совпадениям, затрудняя точное определение их местоположения. Маскировка помогает игнорировать такие участки и сосредоточиться на значимых, уникальных регионах, что важно для поиска мутаций, идентификации генов и анализа регуляторных элементов.
Цель этого и последующих заданий — написать программу для поиска и маскирования повторов в последовательности ДНК.
Задача. Напишите программу, которая принимает строку, состоящую из символов A, T, G и C, и находит самый длинный повтор из одной буквы. Если в строке есть несколько одинаково длинных повторов, нужно вывести первый из них.
Формат входных данных
Программа должна принять (через input()) одну строку ДНК.
Формат выходных данных
Программа должна вывести (через print()) последовательность самого длинного повтора.
Тестовые данные
| Номер теста | Стандартный ввод | Стандартный вывод |
|---|---|---|
1 |
AAATTTGGGCCCCAAGGG |
CCCC |
Ниже представлено решение на языке Python.
# Код решения
def find_longest_single_char_repeat(dna_sequence):
max_repeat = ''
current_char = ''
current_length = 0
max_length = 0
for char in dna_sequence:
if char == current_char:
current_length += 1
else:
current_char = char
current_length = 1
if current_length > max_length:
max_length = current_length
max_repeat = current_char * current_length
return max_repeat
dna_sequence = input()
longest_repeat = find_longest_single_char_repeat(dna_sequence)
print(longest_repeat)
Напишите программу, которая принимает строку, состоящую из символов A, T, G и C, и находит «самый длинный повтор из двух символов», встречающийся подряд более двух раз. Если в строке есть несколько одинаково длинных повторов, выведите первый из них.
Формат входных данных
Программа должна принять (через input()) одну строку ДНК.
Формат выходных данных
Программа должна вывести (через print()) последовательность самого длинного повтора.
Тестовые данные
| Номер теста | Стандартный ввод | Стандартный вывод |
|---|---|---|
1 |
ATATATGCGCGCGTTTTCC |
ATATAT |
Ниже представлено решение на языке Python.
def find_longest_two_letter_repeat(dna):
"""
Находит самый длинный повтор из двух символов, повторяющийся более двух раз подряд.
:param dna: строка ДНК
:return: строка самого длинного повтора
"""
max_length = 0
max_sequence = ""
current_length = 1 # начинаем с 1, так как одна пара уже есть
current_pair = dna[:2] if len(dna) >= 2 else dna # первая пара или первый символ
for i in range(2, len(dna), 2): # идем с шагом 2
pair = dna[i:i + 2] # текущая пара
if pair == current_pair: # если пара совпадает с предыдущей
current_length += 1
else:
# Завершение текущего повтора
repeated_sequence = current_pair * current_length
if len(repeated_sequence) > max_length:
max_length = len(repeated_sequence)
max_sequence = repeated_sequence
# Обновляем текущий повтор
current_pair = pair
current_length = 1
# Обрабатываем последний повтор
repeated_sequence = current_pair * current_length
if len(repeated_sequence) > max_length:
max_length = len(repeated_sequence)
max_sequence = repeated_sequence
# В случае, если строка не делится ровно на пары, проверяем последние символы
if len(dna) % 2 != 0:
last_char_repeat = dna[-1] * dna.count(dna[-1])
if len(last_char_repeat) > max_length:
max_sequence = last_char_repeat
return max_sequence
dna_input = input()
result = find_longest_two_letter_repeat(dna_input)
print(result)
Напишите программу, которая принимает строку, состоящую из символов A, T, G и C, и заменяет в ней все паттерны из одной или двух букв, встречающиеся подряд более двух раз, на соответствующее количество букв N.
Формат входных данных
Программа должна принять (через input()) одну строку ДНК.
Формат выходных данных
Программа должна вывести (через print()) последовательность самого длинного повтора.
Тестовые данные
| Номер теста | Стандартный ввод | Стандартный вывод |
|---|---|---|
1 |
ATATATGCGCGCGTTTTCC |
NNNNNNNNNNNNGNNNNCC |
Ниже представлено решение на языке Python.
import re
def mask_repeats(dna_sequence):
dna_sequence = re.sub(r'(A|T|G|C)\1{2,}', lambda m: 'N' * len(m.group(0)), dna_sequence)
dna_sequence = re.sub(r'(?=(AT|AG|AC|TA|TG|TC|GA|GT|GC|CA|CT|CG))\1\1{2,}', lambda m: 'N' * len(m.group(0)), dna_sequence)
return dna_sequence
dna_input = input("")
masked_dna = mask_repeats(dna_input)
print(masked_dna)
Умение анализировать научную информацию, работать с литературными источниками, письменно излагать свои мысли является крайне востребованным навыком для молодого исследователя. Опыт проведения отборочного тура НТО показал, что написание короткого текста с развернутым ответом на вопрос оказалось одним из самых трудных и интересных заданий.
Нобелевская премия по физиологии и медицине в 2024 году присуждена за открытие микроРНК и их роли в посттранскрипционной регуляции генов.
Задание. Напишите текст (эссе) объемом не более 3000 символов (с учетом пробелов) на тему: «Каким образом микроРНК регулируют экспрессию генов? Какие преимущества имеет данный механизм перед другими способами регуляции?»
Проведите анализ литературных данных. Сопроводите текст ссылками на авторитетные источники. Аргументируйте точку зрения.
- Длина эссе — не более 3000 символов (с учетом пробелов).
- Правильная аргументация, обоснование ответа.
- Отсутствие биологических ошибок.
- Ссылки на литературные источники.
- Текст написан лично автором, отсутствуют заимствованные фрагменты текста.
- Эссе, имеющие явные признаки использования ChatGPT, Алиса ГПТ и подобных инструментов, могут быть оценены в 0 (ноль) баллов.
- В зачет идет одно лучшее эссе от команды.
- При наличии нескольких одинаковых/стереотипных текстов, будет оцениваться текст, загруженный первым, остальные будут оценены в 0 баллов.
- При совпадении фрагментов текстов у разных команд будет оценен первый загруженный текст, остальные будут оценены в 0 баллов.
- При превышении рекомендованной длины текста оцениваться будут первые 3000 символов (с учетом пробелов).
- Список литературы включается (!) в состав 3000 символов.
Командные задачи третьего блока для 8–9 классов открыты для решения. Соревнование доступно на платформе Яндекс.Контест: https://contest.yandex.ru/contest/70125/enter/.
В некоторых случаях бывает очень полезно добавлять dUTP в смесь для ПЦР вместо dTTP. Это позволяет предотвращать перекрестную контаминацию чужими образцами. Проанализируем, что произойдет, если замешать реакционную смесь для ПЦР, содержащую dTTP и dUTP, а потом очистить продукты реакции от «контаминированных».
Для реакции ПЦР использовали мастер-микс 5х, содержащий Taq ДНК-полимеразу с «горячим стартом», смесь dNTP (dATP, dGTP и dCTP — 0,6 мM, dTTP/dUTP — 0,3 мM), 15 мМ ионов \(\ce{Mg^2+}\), реакционный буфер, глицерин, красители (красный и желтый). Замешали реакционную смесь объемом 25 мкл, содержащую 1х мастер-микс, прямые (ATG) и обратные (GCA) праймеры по 0,4 мкМ и ДНК-матрицу: ATGTGCCACGCATGC — 1 копия на мкл реакционной смеси. После трех раундов репликации полученную смесь обработали UDG — урацил-ДНК-гликозилазой, гидролизующей урацил в ДНК.
Определите процент продуктов, подвергшихся гидролизу UDG, если ДНК-полимераза включает dUTP каждый третий комплементарный нуклеотид от начала синтеза новой цепи. В ответе укажите десятичное число с точностью до одного знака после запятой.

Пример ответа: 100,0.
87,5.
Выберите верные утверждения о подборе праймеров для наработки гена с генома для клонирования в экспрессионный плазмидный вектор для продукции белка:
- Праймеры могут отжигаться в любом месте, главное, чтобы последовательность гена находилась между ними.
- Праймеры должны иметь температуры плавления, различающиеся не более, чем на 5 °C.
- Праймеры должны обязательно иметь одинаковую длину — не более 20 нт.
- GC-состав прямого и обратного праймера может отличаться, главное, чтобы он был менее 30%.
- Температура отжига праймера вычисляется по формуле \(T=2(A+T)+4(G+C)\).
- Праймеры должны отжигаться ровно на начало и конец гена.
- GC-состав прямого и обратного праймеров должен находиться в районе 40–60%.
B, F, G.
Основываясь на предыдущем задании, подберите наилучшие праймеры для получения ампликона с гена X для клонирования в экспрессионный вектор для последующей наработки белка.
Последовательность гена X (\(5^\prime\rightarrow3^\prime\)):
Последовательности праймеров (\(5^\prime\rightarrow3^\prime\)):
- TCATGCAGGACACCA.
- TCATGCAGGACAGGC.
- TCATGCAGGACACCATCTAT.
- CTTGTCAGGGCTTGACAC.
- GGTTTACTCGTCCTTCCAT.
- GGAAGGACGAGTAAACCCAC.
- ATGGAAGGACGAGTAAACC.
- ATGGAAGGACGAGTAAAGTT.
- ATAGATGGTGTCCTGCATGA.
- TGCAGGACACCATCTATAC.
С помощью инструмента в программе UGENE проверить каждую из предложенных последовательностей праймеров.
Список литературы
- Возможный инструмент для решения задачи: UGENE https://ugene.net/download-all.html.
3, 7.
Белки — это полипептиды, состоящие из аминокислот. Аминокислоты делятся на типы по характеру их бокового радикала. В зависимости от рН среды раствора полипептид может быть заряжен положительно, отрицательно или нейтрально. В свою очередь, общий заряд полипептида складывается из зарядов остатков аминокислот, входящих в его состав.
Определите направление движения полипептида FENNGY при pH среды, равной 3,9, 6,0 или 10,9. Обратите внимание, что в каноническом виде последовательность аминокислот записывается в направлении от N-конца к С-концу.
Оценить заряд полипептида при различных значениях рН, используя значения pKa каждой функциональной группы.
| рН/направление | К катоду | К аноду |
|---|---|---|
| 3,9 | + | |
| 6,0 | + | |
| 10,9 | + |
В результате воздействия различных внешних и внутренних факторов, например, ультрафиолета или активных форм кислорода, генетический материал клетки нарушает свою структуру. Наиболее часто из-за этого образуются окислительные повреждения ДНК, в частности — 8-оксогуанин. Количество окислительных повреждений в одной клетке за один день может достигать \(10^6\)–\(10^7\). Чтобы нивелировать такое огромное количество повреждений, внутри клетки существует система ферментов эксцизионной репарации оснований (base excision repair — BER). За удаление 8-оксогуанина в человеческих клетках ответственна 8-оксогуанин-ДНК-гликозилаза (hOGG1, h — human).
Как известно, для многих ферментов существуют гомологи из других организмов, что характерно и для hOGG1. В клетках E. coli также присутствует система BER, а ДНК-гликозилаза Fpg (DNA-formamidopyrimedine-glycosylase — формамидо-пиримидин-ДНК-гликозилаза) имеет схожую каталитическую активность, как и hOGG1. Но, что интересно, в отличие от других гомологичных белков hOGG1 и Fpg, имеют совершенно разные аминокислотные последовательности как в первичной, вторичной, так и в третичной структуре!
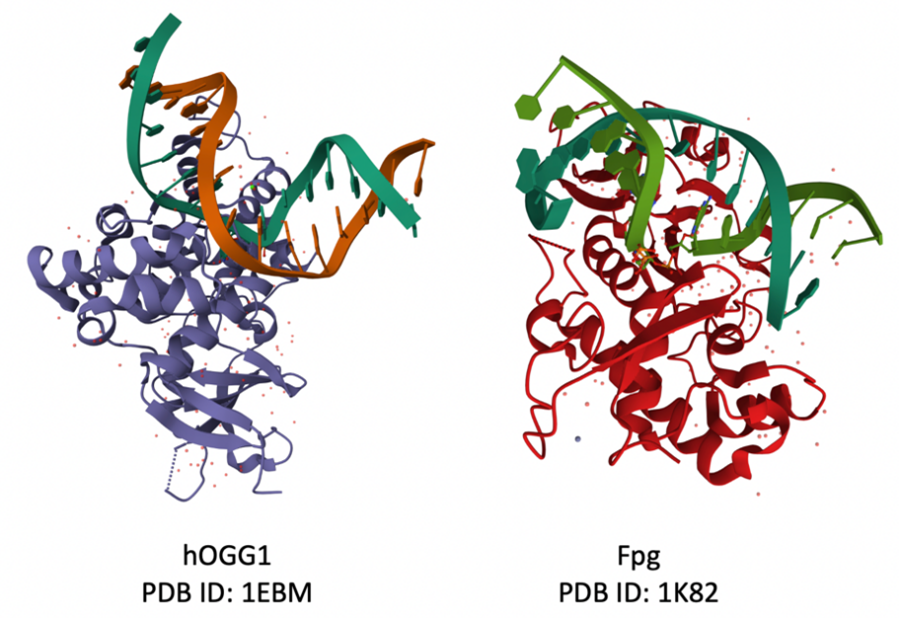
Таким образом, данные ферменты гомологичны по функциям, но негомологичны по строению. Чтобы частично продемонстрировать это, проведем следующий эксперимент.
Для сравнения первичной структуры ферментов hOGG1 и Fpg их независимо обработали бромцианом и проанализировали продукты реакции методом белкового гель-электрофореза. Определите длины самых больших продуктов в каждой реакции, если бромциан расщепляет пептидные связи, образованные Met. В ответе укажите абсолютную разницу (модуль) между этими длинами.
Список литературы
- Ссылка на последовательность OGG1 (https://www.ncbi.nlm.nih.gov/nuccore/U88527): send to \(\Rightarrow\) Coding Sequences \(\Rightarrow\) Create file.
- Ссылка на последовательность Fpg (https://www.ncbi.nlm.nih.gov/nuccore/NC_000913.3): send to \(\Rightarrow\) Coding Sequences \(\Rightarrow\) Create file.
- Возможный инструмент для решения задачи: UGENE https://ugene.net/download-all.html.
37.
Плазмиду pBlueScript SK(–), содержащую ген бета-галоктозидазы 11 из Arabidopsis Thaliana, получали методом клонирования по липким концам при помощи рестриктаз SpeI с 5\(^\prime\)-конца и XbaI с 3\(^\prime\)-конца.
Затем было необходимо переклонировать вставку BGAL11 в другой плазмидный вектор по сайтам рестрикции EcoRI и XbaI. Для этого замешали реакционную смесь объемом 50 мкл, содержащую 2024 нг плазмиды со вставкой, 2 ЕА (единиц активности) XbaI и 2 ЕА EcoRI, а также реакционный буфер, в котором активность XbaI составляет 100%, а EcoRI — 75% (ожидается неполная рестрикция). Реакционную смесь инкубировали при 37 °C в течение 1 ч. После чего инактивировали ферменты при 65 °C в течение 20 мин и анализировали в агарозном геле.
Определите минимальный объем реакционной смеси, который необходимо нанести на 1%-й агарозный гель, чтобы визуализировать вставку.
Очень важно правильно подобрать правильное количество ДНК для нанесения на агарозный гель, от этого зависит степень визуализации и верная интерпретация результатов рестрикции. При «перегрузе» (слишком большом количестве нанесенной ДНК) будет сложно разделить продукты схожей длины. Кроме того, визуализация при помощи бромистого этидия имеет чувствительность 50 нг ДНК на дорожку, меньшее количество для фрагментов короче 50 пн будет невозможно задетектировать.
В ответе укажите объем реакционной смеси в мкл в виде целого натурального числа (при необходимости округлите до ближайшего целого числа).
- Для получения плазмиды pBlueScript SK(–) со вставкой гена BGAL11 необходимо скачать последовательности и открыть их в UGENE. Затем в начало и конец гена BGAL11 добавить последовательности соответствующих рестриктаз. Чтобы добавить последовательность, воспользуйтесь методом «редактирование» \(\Rightarrow\) «вставить участок».
- В результате рестрикции in silico суммарная длина продуктов будет отличаться от длины вектора. Это связано с тем, что UGENE не учитывает длину оверхенгов (липких концов). На ответ задачи это не влияет в связи с округлением.
Описано в комментарии к задаче.
Список литературы
- Ссылка на плазмиду pBlueScript SK(–) (https://www.addgene.org/vector-database/1952/): copy sequence \(\Rightarrow\) UGENE создать новую последовательность.
- Ссылка на последовательность гена BGAL11 (https://www.ncbi.nlm.nih.gov/nuccore/NC_003075.7): send to \(\Rightarrow\) Coding Sequences \(\Rightarrow\) Create file.
- Возможный инструмент для решения задачи: UGENE https://ugene.net/download-all.html.
4.
В биохимии и молекулярной биологии очень важно измерять различные кинетические параметры ферментативных реакций. Это необходимо не только для характеризации фермента и биохимических процессов, но и для продвижения фармакологических технологий.
К одной из наиболее известных и простых моделей ферментативной кинетики можно отнести модель Михаэлиса – Ментен, разработанную в 1913 г. Она объясняет характерную гиперболическую зависимость активности фермента от концентрации субстрата и позволяет получать константы, которые количественно характеризуют эффективность фермента. Простейшая кинетическая схема, для которой справедливо уравнение Михаэлиса – Ментен: \[\ce{E + S \Leftrightarrow ES -> E + P},\] где E — фермент, S — субстрат, ES — комплекс фермент-субстрат, P — продукт.
Кинетическое уравнение имеет следующий вид: \[v= \frac{V_m \cdot S}{S + K_m},\] где \(v\) — скорость реакции, \(V_m\) — максимальная скорость реакции, \(K_m\) — константа Михаэлиса (численно равна концентрации субстрата, при которой скорость реакции составляет половину от максимальной), \(S\) — концентрация субстрата.
Константа \(K_m\) показывает сродство фермента к субстрату, т. е. насколько хорошо фермент связывает субстрат. Как известно, для ДНК-полимераз субстратами выступают ДНК-матрица и dNTP, поэтому попробуем оценить сродство ДНК-полимеразы к dNTP.
Для ферментативной реакции включения dCTP напротив гуанина на субстрате типа «праймер-матрица» фрагментом Кленова ДНК-полимеразы I E. coli измерили кинетические параметры в условиях кинетики Михаэлиса – Ментен. Скорость реакции при концентрации субстрата 100 нМ составила 8,54 пМ/с, а максимальная скорость реакции оказалась равной 0,035 нМ/с. Рассчитайте константу Михаэлиса. В ответе укажите число с точностью до двух знаков после запятой и размерностью мкМ.
Пример ответа: 12,34.
Список литературы
- Статья в Wikipedia про уравнение Михаэлиса – Ментен: https://ru.wikipedia.org/wiki/Уравнение_Михаэлиса_—_Ментен
0,31.
На долю лаборанта часто выпадают непростые задачи, с которыми не любят возиться более взрослые сотрудники, вот и в этот раз лаборанта Василия подозвал к себе научный руководитель, чтобы поручить сверхважное задание. Лаборатория профессора Живодерова занимается изучением гена BRCA 1 (BReast CAncer gene 1), который кодирует белок, участвующий в репарации ДНК и поддержании стабильности генома.
Чтобы лучше изучить его, сотрудники лаборатории вместе с лаборантом Василием применяют ДНК-зонды, которые за счет комплементарности могут гибридизоваться на последовательности участков интересующего гена. Сейчас Василию необходимо гибридизовать зонд со вторым экзоном изучаемого гена (зонд полностью покрывает искомый экзон). Для того чтобы правильно поставить реакцию, необходимо все предварительно рассчитать. Одним из важнейших параметров является концентрация зонда — ее и нужно посчитать.
Задача. Рассчитать концентрацию зонда, выраженную в пмоль/мкл. Для этого придется воспользоваться следующей формулой: \[C(\text{пмоль/мкл}) = \frac{OD \cdot 100}{1{,}5 \cdot N_A + 0{,}71 \cdot N_C + 1{,}2 \cdot N_G + 0{,}84 \cdot N_T},\] где \(N_A\), \(N_C\), \(N_G\), \(N_T\) — количество соответствующего основания в олигонуклеотиде, \(OD\) — оптическая плотность при 260 нм, выраженная в ОЕ/мл (принять равной 2).
Для того чтобы посчитать количество каждого нуклеотида в зонде, необходимо найти в NCBI ген BRCA 1, у него определить транскрипт с идентификатором NM_001407593.1. В нем нужно будет найти второй экзон, который соответствует последовательности зонда. Помните, что зонд покрывает весь экзон и комплементарен ему!
Ответ запишите, округлив до сотых. В качестве десятичного разделителя используйте точку (например, 1.23).
3.44.
Позиционная весовая матрица (PWM, Position Weight Matrix) — это способ описания важных участков последовательностей ДНК, например, сайта связывания белков. В каждой позиции матрица показывает вероятность нахождения одного из нуклеотидов: A, C, G или T. Если в какой-то позиции всегда встречается один и тот же нуклеотид, то вероятность для него будет близка к 1, а для остальных — к 0. Если нуклеотиды распределены равномерно, вероятности будут примерно одинаковыми. PWM часто используют в биоинформатике для поиска биологически значимых участков в последовательности ДНК.
Например, белки, такие как транскрипционные факторы, связываются с определенными мотивами ДНК, и PWM помогает предсказать, где такие мотивы могут находиться. Матрица строится на основе множества выровненных последовательностей, которые содержат этот мотив. PWM полезна тем, что учитывает вариабельность последовательностей: даже если мотив немного отличается в разных местах, его все равно можно обнаружить благодаря весам в матрице. Цель этой задачи — написать программу для построения позиционной весовой матрицы.
Задача. Напишите программу, которая будет принимать на вход выровненные последовательности ДНК и строить для них позиционную весовую матрицу (PWM). Для того чтобы построить PWM, посчитайте частоту каждого нуклеотида в каждой позиции и разделите это число на общее количество последовательностей, чтобы получить вероятности. В результате должна получиться матрица \(N \times M\), где \(N\) — количество нуклеотидов (4), а \(M\) — количество позиций в последовательностях. В ответ выведите список из четырех перечней, где каждый вложенный список — частоты нуклеотидов A, C, G, T в каждой позиции в последовательности.
Формат входных данных
Программа должна принять (через input()) строку, содержащую произвольное количество последовательностей ДНК (больше двух), разделенных пробелами.
Формат выходных данных
Программа должна вывести (через print()) позиционную весовую матрицу в виде вложенных списков.
Тестовые данные
| Номер теста | Стандартный ввод | Стандартный вывод |
|---|---|---|
1 |
ACGTA GCTAG GCCTA GAATC |
[[0.25, 0.25, 0.25, 0.25, 0.5], [0.0, 0.75, 0.25, 0.0, 0.25], , [0.0, 0.0, 0.25, 0.75, 0.0]] |
Ниже представлено решение на языке Python.
def build_pwm(sequences):
# Инициализация матрицы для подсчета частот
pwm = [[0] * len(sequences[0]) for _ in range(4)]
nucleotide_index = {'A': 0, 'C': 1, 'G': 2, 'T': 3}
# Подсчет частоты нуклеотидов
for seq in sequences:
for i, nucleotide in enumerate(seq):
pwm[nucleotide_index[nucleotide]][i] += 1
# Количество последовательностей
num_sequences = len(sequences)
# Преобразование частот в вероятности
for i in range(4):
for j in range(len(pwm[i])):
pwm[i][j] /= num_sequences
return pwm
input_sequences = input()
sequences = input_sequences.split()
pwmmatrix = build_pwm(sequences)
print(pwmmatrix)
Напишите программу, которая будет принимать на вход выровненные последовательности ДНК (разделенные пробелом) для построения PWM, и через запятую — целевую последовательность ДНК, в которой нужно выполнять поиск мотива. Сначала программа должна построить PWM для последовательностей, перечисленные до запятой. Затем необходимо найти в целевой последовательности ДНК подпоследовательность, максимально соответствующую PWM (имеет наибольшую вероятность). Если таких подпоследовательностей в целевой последовательности несколько, приведите первую из них (ту, которая находится ближе к началу последовательности).
Формат входных данных
Программа должна принять (через input()) строку, содержащую произвольное количество последовательностей ДНК (больше двух), разделенных пробелами и целевую последовательность ДНК, отделенную от первых запятой.
Формат выходных данных
Программа должна вывести (через print()) позиционную весовую матрицу в виде вложенных списков.
Тестовые данные
| Номер теста | Стандартный ввод | Стандартный вывод |
|---|---|---|
1 |
AAGG TTGC ACGG TTCG, ACGGTTGGAAGG |
TTGG |
Ниже представлено решение на языке Python.
def build_pwm(sequences):
# Определяем длину последовательностей
seqlength = len(sequences[0])
pwm = []
# Инициализируем матрицу нулями
for _ in range(seqlength):
pwm.append({'A': 0, 'C': 0, 'G': 0, 'T': 0})
# Заполняем матрицу
for seq in sequences:
for i, nucleotide in enumerate(seq):
pwm[i][nucleotide] += 1
# Преобразуем в вероятности
for i in range(seqlength):
total = sum(pwm[i].values())
for nucleotide in pwm[i]:
pwm[i][nucleotide] /= total
return pwm
def findbestmatch(pwm, targetsequence):
best_score = -1
best_subsequence = ""
pwmlength = len(pwm)
for i in range(len(targetsequence) - pwmlength + 1):
subsequence = targetsequence[i:i + pwmlength]
score = 1
for j, nucleotide in enumerate(subsequence):
score *= pwm[j][nucleotide]
if score > best_score:
best_score = score
best_subsequence = subsequence
return best_subsequence
input_data = input()
sequencespart, targetsequence = input_data.split(',')
sequences = sequencespart.strip().split()
pwm = build_pwm(sequences)
bestmatch = findbestmatch(pwm, targetsequence.strip())
print(bestmatch)
Три студента-биотехнолога — Антон, Борис и Василий — пытались клонировать последовательность неизвестного гена NOIDEA в плазмидный вектор pUC19, чтобы изучить его фунцию. Они использовали одинаковые образцы вектора и вставки, одни и те же эндонуклеазы рестрикции (EcoRI и BamHI), но разные протоколы обработки фрагментов ДНК перед лигированием:
- Антон обработал щелочной фосфатазой (AP) и вектор, и вставку перед лигированием.
- Борис обработал щелочной фосфатазой только вектор.
- Василий не использовал фосфатазу вообще.
После трансформации компетентных клеток E. coli продуктами лигирования и высева на чашки с ампициллином (маркер устойчивости вектора pUC19) только у одного из студентов выросли колонии, содержащие плазмиды с правильно встроенным геном. У кого из студентов получился успешный эксперимент?
Варианты ответа:
- Антон,
- Борис,
- Василий.
2.
В ходе рутинных генно-инженерных манипуляций плазмида размером 9000 пар азотистых оснований была разрезана эндонуклеазами рестрикции на два фрагмента ДНК. Более крупный фрагмент оказался ровно в два раза больше, чем меньший фрагмент. Меньший фрагмент был выделен и очищен из агарозного геля после электрофоретического разделения.
Задача. Какое количество рибонуклеотидов содержится в более длинном фрагменте ДНК, элюированном из геля?
ДНК не содержит рибонуклеотиды, но дезоксирибонуклеотиды.
0.
Пандемия коронавирусной инфекции стала трудным испытанием для системы здравоохранения. Компании рынка «Хелснет» НТИ внесли значительный вклад в решение задач по ПЦР- и ИФА-диагностике коронавирусной инфекции, разработке новых вакцин. Большую роль в этом сыграли не только специалисты, работающие в молекулярно-биологических лабораториях, но и биоинформатики.
Сквозная технология Национальной технологической инициативы (НТИ) «Управление свойствами биологических объектов» основана на достижениях инженерной биологии: генетической инженерии, биотехнологии, молекулярной биологии, биохимии и многих других. Актуальность исследований в области разработки новых инструментов генетической инженерии и геномного редактирования сегодня не вызывает сомнений. Большая часть новых продуктов фармацевтической индустрии, биотехнологий и других отраслей основана на использовании инструментов, созданных в конце XX века (генетическая инженерия), сегодня активно разрабатывают и другие подходы, среди которых особое место занимает синтетическая биология и технология геномного редактирования.
Командные задачи первого блока для 10–11 классов открыты для решения. Соревнование доступно на платформе Яндекс.Контест: https://contest.yandex.ru/contest/70126/enter/.
Гель-электрофорез — это метод, используемый для разделения фрагментов ДНК (или других макромолекул, например, РНК или белков) в зависимости от их размера и заряда. Электрофорез — это процесс пропускания тока через гель, содержащий интересующие нас молекулы.

Какие суждения справедливы для фракционирования ДНК-методом гель-электрофореза? Выберите верные суждения.
- ДНК заряжена отрицательно и мигрирует от катода к аноду.
- Более короткие фрагменты ДНК имеют меньший удельный заряд и двигаются медленнее.
- Агароза или полиакриламид необходимы для того, чтобы отделить более короткие молекулы от более длинных.
- Для того чтобы обеспечить заряд ДНК, в буферный раствор для нанесения добавляют додецилсульфат натрия (SDS).
- Разделение ДНК проводят в дистиллированной воде с низкой электропроводностью.
A, C.
Репликация ДНК — процесс образования из молекулы ДНК двух ее дочерних «копий». Это довольно сложный процесс, в котором задействовано порядка 20 различных ферментов, и каждый из них выполняет свою функцию. Репликация начинается на сайте инициализации с расплетения двойной спирали, и при этом образуется репликационная вилка, где и происходит синтез комплементарных цепей.
Какие суждения характерны для репликационной вилки эукариот?
Выберите верные варианты ответа:
- Репликацию основной цепи осуществляет ДНК-полимераза эпсилон.
- Хеликаза CMG находится и на лидирующей, и на отстающей цепи.
- ДНК-полимеразы, реплицирующие основною и отстающую цепи, связаны вместе комплексом скользящей застежки (PCNA).
- Фрагменты Оказаки лигируются ДНК-полимеразой альфа.
- В репликации отстающей цепи принимает участие ДНК-полимераза альфа.
A, E.
Долгое время ученые, изучавшие репликацию ДНК, не могли понять, каким образом обе цепи ДНК могут реплицироваться одновременно. Ведь они антипараллельны, а ДНК-полимераза может присоединять нуклеотиды только к 3\(^\prime\)-концу, а значит, она может удлинять только одну из двух растущих цепей!
На этот вопрос смог ответить Рейджи Оказаки, который провел ряд экспериментов и обнаружил, что при репликации у бактерий большая часть новообразованной ДНК обнаруживается в форме небольших кусков, названными фрагментами Оказаки.

Какой фермент принимает участие в удалении РНК-праймера между фрагментами Окадзаки?
Выберите верные варианты ответа:
- ДНК-лигаза.
- ДНК-полимераза I у E. coli.
- ДНК-полимераза альфа у эукариот.
- РНКаза H у эукариот.
- Хеликаза у эукариот.
B, D.
В лаборатории для многих целей используются центрифуги или микроспины (microspin). Принцип их действия основан на центробежной силе, что позволяет осаждать раствор в пробирке или разделять компоненты раствора по массе (например, клетки от среды для культивирования). Для того чтобы эти приборы работали исправно, при расстановке пробирок нужно следить за тем, чтобы они были в равновесии.
На рис. 2.4 можно увидеть полностью загруженный микроспин на 12 положений.

Необходимо разместить в этом приборе пять пробирок одинаковой массы таким образом, чтобы он находился в равновесии. Выберите из предложенных вариантов номера положений, в которых можно разместить пробирки без угрозы. Для удобства будем вести отсчет положений от помеченного стрелкой и по часовой стрелке.
- 2, 6, 10, 7, 1.
- 1, 3, 6, 9, 12.
- 4, 12, 8, 3, 9.
- 6, 7, 9, 2, 10.
- Нужна еще одна пробирка!
A, C.
Одним из самых популярных и используемых методов секвенирования ДНК долгое время был метод Сэнгера, за который в 1980 году была присуждена Нобелевская премия. Этот метод позволяет считывать последовательности до 1000 пар нуклеотидов, и потому активно используется для секвенирования и по сей день, например, отдельных участков генома для анализа мутаций.
При проведении секвенирования по Сэнгеру реакционную смесь разделяют методом капиллярного гель-электрофореза. На рис. 2.5 приведена «секвенирующая лестница», по которой можно определить последовательность ДНК.

Изучите рис. 2.5 и рассчитайте, какое количество энергии необходимо затратить, чтобы разорвать дуплекс из расшифрованной и комплементарной цепей, если энергия одной водородной связи равна в среднем 40 кДж/моль. В ответе укажите целое натуральное число без размерности.
Список литературы
2400.
Научный руководитель выдал лаборанту пробирку с плазмидой pcDNA3.2_YFP, с помощью которой можно нарабатывать желтый флуоресцентный белок (англ. Yellow Fluorescent Protein, YFP) в клетках млекопитающих, и попросил убедиться в ее соответствии коммерческой плазмиде с помощью построения рестрикционной карты на основе следующих ферментов Apa I, BciV I, Bts I, Dra III и Nco I.
Он целый день проводил реакции рестрикции в различных вариантах плазмиды pcDNA3.2_YEF и попросил напарника провести гель-электрофорез вместо себя. Напарник выполнил просьбу, но случайно забыл, в каком порядке наносил образцы на агарозный гель. Фотография гель-электрофореза представлена на рис. 2.6.
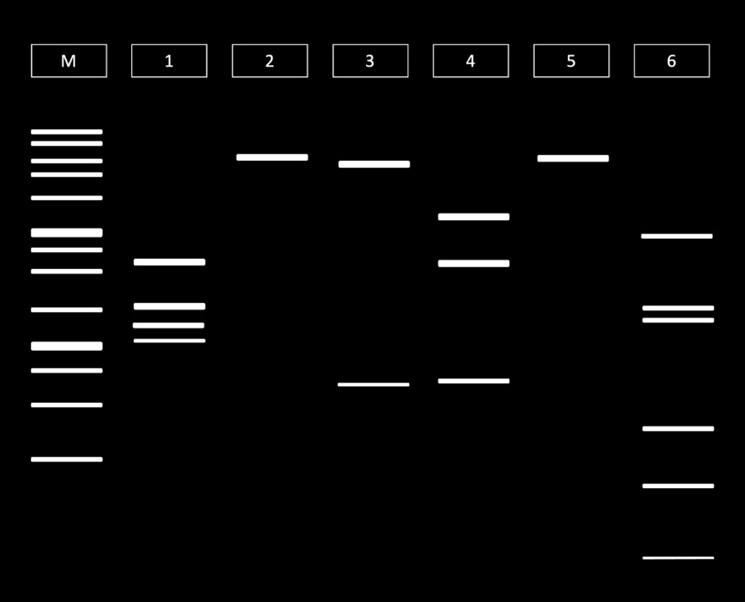
Выберите варианты ответов, где в реакционной смеси присутствуют продукты гидролиза плазмиды pcDNA3.2_YFP, рестриктазами Apa I и Dra III, если в качестве ДНК-маркера использовалась смесь 1kb «СибЭнзайм».
- Дорожка 1.
- Дорожка 2.
- Дорожка 3.
- Дорожка 4.
- Дорожка 5.
- Дорожка 6.
- Представлены результаты рестрикции другой плазмиды.
- Представлены результаты рестрикции другими рестриктазами.
Список литературы
- Карта плазмиды pcDNA3.2_YFP: https://www.addgene.org/84910/ (дополнительная ссылка на карту https://disk.yandex.ru/i/HllW4S6D7t2KzA).
- ДНК-маркер СибЭнзайм: https://sibenzyme.com/product/1-kb-13-fragmentov-ot-0-25-do-10-kb/ (pdf-версия веб-страницы https://disk.yandex.ru/i/pcUDr4DL5mLi2Q).
- Предлагаемый софт для решения задачи: UGENE http://ugene.net/ru/.
B, C, E.
Белок PCNA играет исключительно важную роль в репликации ДНК эукариот и, например, связывает ДНК-полимеразу дельта (эпсилон) при репликации основной (отстающей) цепи.
Изучите информацию о гене PCNA человека https://www.ncbi.nlm.nih.gov/gene?cmd=retrieve&dopt=default&rn=1&list_uids=5111 (pdf-версия веб-страницы https://disk.yandex.ru/i/JFHWzeUK2s9Cyw) в базе данных NCBI.
Выберите верные варианты ответа:
- Ген PCNA человека лежит в 20 хромосоме.
- Ген PCNA человека лежит в 12 хромосоме.
- Ген PCNA человека лежит в 7 хромосоме.
- Ген PCNA человека содержит 20 экзонов.
- Ген PCNA человека содержит 12 экзонов.
- Ген PCNA человека содержит 7 экзонов.
A, F.
Белок PCNA играет исключительно важную роль в репликации ДНК эукариот. Изучите информацию о гене PCNA человека в genbank https://www.ncbi.nlm.nih.gov/nuccore/NG_047066.1 (pdf-версия веб-страницы https://disk.yandex.ru/i/Ks_X3BPhdeH87g). Определите число аминокислотных остатков в данном белке, исходя из последовательности CDS.
261.
Откройте аминокислотную последовательность PCNA из прошлой задачи в UGENE и определите молекулярную массу белка.
Введите молекулярную массу PCNA в грамм на моль (г/моль) в виде целого числа.
28772.
Командные задачи второго блока для 10–11 классов открыты для решения. Соревнование доступно на платформе Яндекс.Контест: https://contest.yandex.ru/contest/70127/enter/.
Мутации в генах могут приводить к развитию различных заболеваний. Одной из таких болезней является серповидно-клеточная анемия, вызванная точечной мутацией в гене гемоглобина.
Задача. Найдите мутацию, вызывающую серповидно-клеточную анемию, и проведите анализ последствий этой мутации.
Поиск мутации: найдите последовательность нормального гена гемоглобина и мутантного гена, вызвавшего серповидноклеточную анемию. Используйте базу данных OMIM (Online Mendelian Inheritance in Man) для поиска информации о мутации.
Выравнивание последовательностей: проведите выравнивание нормальной и мутированной последовательностей с помощью программы Clustal Omega или аналогичных инструментов.
Найдите аминокислоту, замена которой приводит к развитию серповидноклеотчной анемии. В ответ введите эту аминокислоту (из нормального белка) и по одной фланкирующей аминокислоте слева и справа (в трехбуквенном коде, с большой буквы). Пример: GlyAspTrp.
ProGluGlu.
Шпилька в последовательности ДНК — это структура, образуемая двумя строго комплементарными участками одинаковой длины, которые расположены зеркально относительно середины участка. Учитывать шпильки в последовательности ДНК необходимо при дизайне различных олигонуклеотидов, в том числе праймеров.
Задача. Напишите программу, которая будет осуществлять поиск шпильки в последовательности ДНК. Шпилькой считается последовательность, состоящая из двух комплементарных половин без петли. В данной задаче необходимо считать, что минимальная длина шпильки — 4 нуклеотида. В последовательности может не быть шпильки, или вся последовательность может представлять собой шпильку.
Формат входных данных
Программа должна принять (через input()) одну строку ДНК.
Формат выходных данных
Программа должна вывести (через print()) последовательность с маскированными повторами.
Тестовые данные
| Номер теста | Стандартный ввод | Стандартный вывод |
|---|---|---|
1 |
ACTGCGCCAG |
GCGC |
Ниже представлено решение на языке Python.
a = input().upper()
maxi = 0
p = '' # Инициализируем p как пустую строку
for i in range(len(a)):
for j in range(i + 1, len(a)):
srez = a[i:j + 1] # Исправлено: srez должен быть присвоен, а не a[i:j+1]
if len(srez) < 4:
continue
if len(srez) % 2 == 0:
flag = True
a1 = srez[:len(srez) // 2]
a2 = srez[len(srez) // 2:][::-1]
for k in range(len(a1)):
if (a1[k] == 'C' and a2[k] == 'G') or \
(a1[k] == 'T' and a2[k] == 'A') or \
(a1[k] == 'G' and a2[k] == 'C') or \
(a1[k] == 'A' and a2[k] == 'T'):
pass
else:
flag = False
break
if flag == True:
if len(srez) > maxi:
maxi = len(srez)
p = srez
print(p)
Поиск самой длинной общей подстроки (LCS) — это задача нахождения наибольшего непрерывного совпадения между двумя последовательностями. LCS часто используют в биоинформатике для поиска консервативных регионов, которые могут быть полезны при дизайне тест-систем.
Например, при создании теста для вирусов гриппа A и B можно искать общую подстроку в генах гемагглютинина (HA), чтобы разработать универсальные праймеры. LCS подходит лучше, чем выравнивание, если требуется строгое совпадение без пропусков, как в случае создания праймеров для ПЦР. Кроме того, поиск LCS быстрее, чем полное выравнивание, что важно для анализа больших данных. В данной задаче необходимо найти самую длинную общую подстроку для двух последовательностей ДНК.
Задача. Напишите программу, которая будет выполнять поиск самой длинной общей подстроки в двух последовательностях ДНК.
Формат входных данных
Программа должна принять (через input()) строку, содержащую две последовательности ДНК, разделенные пробелом.
Формат выходных данных
Программа должна вывести (через print()) последовательность самой длинной общей подстроки для этих двух последовательностей.
Тестовые данные
| Номер теста | Стандартный ввод | Стандартный вывод |
|---|---|---|
1 |
ACCGTT CCAGCA |
CC |
Ниже представлено решение на языке Python.
def longest_common_substring(str1, str2):
substrings = []
for i in range(len(str1)):
for j in range(i + 1, len(str1) + 1):
substrings.append(str1[i:j])
substrings.sort(key=len, reverse=True)
for substr in substrings:
if substr in str2:
return substr
return ""
str1, str2 = input().split()
result = longest_common_substring(str1, str2)
print(result)
Другим методом секвенирования первого поколения было секвенирование Максама – Гилберта. Однако этот метод «вышел из моды», так как в нем используют опасные химикаты, а отсеквенировать длинный фрагмент ДНК невозможно.

Метод Максама – Гилберта основан на внесении разрывов по разным типам азотистых оснований с помощью химического воздействия. После обработки полученные фрагменты визуализируют с помощью денатурирующего полиакриламидного геля.
На рис. 2.7 приведен схематичный результат секвенирования. На первой дорожке образец обрабатывали концентрированной муравьиной кислотой с пиперидином, на второй — диметилсульфатом, на третьей — гидратом гидразина, а на четвертой — смесью гидрата гидразина с пиперидином.
Расшифруйте последовательность. В ответ запишите фрагмент в направлении \(5^\prime\rightarrow3^\prime\) (заглавными латинскими буквами). Пример ответа: ATGC.
GTGTCAGATGATCACTGACTAGG.
В 1980-х годах ученые Марио Капекки, Оливер Смитис и Мартин Эванс разработали метод нокаута (knockout) гена, который позволил продвинуть развитие обратной генетики. В 2007 году эти ученые получили Нобелевскую премию по физиологии и медицине за данный метод.

Одним из главных преимуществ нокаутов генов является то, что они позволяют исследователям изучать функцию конкретного гена in vivo и понимать роль гена в нормальном развитии и физиологии, а также в патологии заболеваний. Сравнивая фенотип организма с нокаутированным геном, исследователи могут получить представление о биологических процессах, в которых участвует ген. Для получения нокаутов генов чаще всего используют систему CRISPR/Cas9, поскольку она наиболее точна, проста в дизайне и использовании по сравнению с другими системами генетического редактирования (TALEN, цинковыми пальцами и др.).
Для изучения некоего гена «X» методом обратной генетики было решено получить его нокаут при помощи белка Cas9, выделенного из Streptococcus pyogenes. Для редактирования была выбрана следующая область гена: 5\(^\prime\)-TAGGAACTAGGCTACGGTCTTTTGCGAACCCTTCGGCTGTATCTAGAGTGCTATTACTACAGTTCACATGGTTCTAGCCTTTATGATTACGGTCCGTCG-3\(^\prime\).
И сконструирована направляющая gRNA со спейсером: UGCUAUUACUACAGUUCACA.
Определите нуклеотидную последовательность PAM и введите ее в направлении \(5^\prime\rightarrow3^\prime\).
TGG.
Процесс нокаута генов с помощью CRISPR/Cas9 включает в себя три основных шага:
- Дизайн направляющей РНК (gRNA, guide — направляющая), которая нацелена на определенное место в геноме.
- Доставка gRNA и фермента Cas9 (который действует как молекулярные ножницы) в целевую клетку.
Восстановление двуцепочечного разрыва в ДНК. Когда клетка репарирует двуцепочечный разрыв, возможно два пути репарации:
- NHEJ (non-homologous end joining, негомологичное соединение концов), в результате которого часто возникают инделы (инсерции или делеции нуклеотидов), что сдвигает открытую рамку считывания;
- HDR (homology directed repair, гомологичная направленная репарация), в результате которой восстановление разрыва осуществляется за счет гомологичной рекомбинации по матрице-донору (как правило, это олигонуклеотид, содержащий нужную мутацию). Если происходит замена большого участка или всего гена, то этот процесс называется нок-ином (knock-in).

Разработанную систему для нокаута гена «X» из прошлой задачи ввели в клетки мышей четырех линий и секвенировали целевую последовательность ДНК после редактирования. Затем в те же клеточные линии ввели ту же систему, но совместно с донором 5\(^\prime\)-TCTAGAGTGCTAATAATAAAGTTCACATGGT-3\(^\prime\), и также секвенировали.
Изучите результаты секвенирования двух экспериментов и определите последовательности с нокаутом гена «X».
- TAGGAACTAGGCTACGGTCTTTTGCGAACCCTTCGGCTGTATCTAGAGTGCTATTACTACAGTTCGACACATGGTTCTAGCCTTTATGATTACGGTCCGTCG.
- TAGGAACTAGGCTACGGTCTTTTGCGAACCCTTCGGCTGTATCTAGAGTGCTATTACTACAGTACATGGTTCTAGCCTTTATGATTACGGTCCGTCG.
- TAGGAACTAGGCTACGGTCTTTTGCGAACCCTTCGGCTGTATCTAGAGTGCTATTACTACAGTTCGACATGGTTCTAGCCTTTATGATTACGGTCCGTCG.
- TAGGAACTAGGCTACGGTCTTTTGCGAACCCTTCGGCTGTATCTAGAGTGCTATTACTACAGACATGGTTCTAGCCTTTATGATTACGGTCCGTCG.
- TAGGAACTAGGCTACGGTCTTTTGCGAACCCTTCGGCTGTATCTAGAGTGCTATTACTACAGTTCACATGGTTCTAGCCTTTATGATTACGGTCCGTCG.
- TAGGAACTAGGCTACGGTCTTTTGCGAACCCTTCGGCTGTATCTAGAGTGCTATTACTACAGTTCACATGGTTCTAGCCTTTATGATTACGGTCCGTCG.
- TAGGAACTAGGCTACGGTCTTTTGCGAACCCTTCGGCTGTATCTAGAGTGCTAATAAAGTTCACATGGTTCTAGCCTTTATGATTACGGTCCGTCG.
- TAGGAACTAGGCTACGGTCTTTTGCGAACCCTTCGGCTGTATCTAGAGTGCTAATAATAAAGTTCACATGGTTCTAGCCTTTATGATTACGGTCCGTCG.
2, 3, 7, 8.
Плазмиду pUC19 (2686 пн) гидролизовали рестриктазой AceIII до полного расщепления. Известно, что реакционная смесь объемом 60 мкл содержала 2024 нг плазмиды.
Задача. Определите минимальный объем реакционной смеси, который необходимо нанести на 1%-й агарозный гель, чтобы визуализировать самый короткий фрагмент. Очень важно правильно подобрать правильное количество ДНК для нанесения на агарозный гель, от этого зависит степень визуализации и верная интерпретация результатов рестрикции. При «перегрузе» (слишком большом количестве нанесенной ДНК) будет сложно разделить продукты схожей длины. Кроме того, визуализация при помощи бромистого этидия имеет чувствительность 50 нг ДНК на дорожку, меньшее количество для фрагментов короче 50 пн будет невозможно задетектировать.
В ответе укажите объем реакционной смеси в мкл в виде целого натурального числа (при необходимости округлите до ближайшего целого числа).
6.
Цикл трикарбоновых кислот (цикл Кребса) — это ключевой этап клеточного окисления глюкозы и других питательных веществ для получения энергии.
В начале цикла трикарбоновых кислот ацетил-КоА, который образуется при окислении глюкозы во время гликолиза, жирных кислот или аминокислот, отдает свою ацетильную группу четырехуглеродному соединению — оксалоацетату (щавелевоуксусной кислоте — ЩУКа), при этом образуется шестиуглеродный цитрат. Цитрат затем изомеризуется в изоцитрат, который далее дегидрируется и декарбоксилируется до пятиуглеродной кислоты — \(\alpha\)-кетоглутарата. \(\alpha\)-кетоглутарат вновь декарбоксилируется, превращаясь в четырехуглеродный сукцинат. Сукцинат затем превращается в фумарат под воздействием сукцинатдегидрогиназы, после чего он в два этапа ферментативно преобразуется снова в оксалоацетат — ЩУКу.

Сукцинатдегидрогеназа (SDH) является центральным звеном между энергетическим обменом (цикл Кребса) и дыхательной цепью, обеспечивая эффективное использование выделенной энергии для синтеза АТФ. SDH состоит из четырех субъединиц: SDHA, SDHB, SDHC и SDHD. Субъединица А содержит ФАД и сайт связывания сукцината, что крайне важно для ферментативного преобразования сукцината в фумарат.
Всем известно, что цикл трикарбоновых кислот представляет собой один из основных энергетических путей в клетках не только животных, но и растений, и грибов, и бактерий. В связи с этим проанализируем генетические особенности субъединицы А сукцинатдегидрогеназы (SDH) у ЛЕБЕДЯ-шипуна (Cygnus olor).
Задача. Используя базу данных NCBI, определите, на какой хромосоме расположен ген SDHA у лебедя-шипуна, количество интронов в кодирующей последовательности гена SDHA и длину продукта трансляции SDHA. Ответ запишите в формате: [№ хромосомы][количество интронов][длина белка]. Пример ответа: 1_2_345.
2_14_661.
Когда механизмы регуляции таких путей, как цикл трикарбоновых кислот, нарушаются, в результате может развиться серьезное заболевание. Ферменты цикла кодируются генами домашнего хозяйства, и отсутствие функциональных копий этих генов может быть фатальным. Мутации генов сукцинатдегидрогеназы (SDH) вызывают РАК надпочечников (феохромоцитому). В культурах клеток с такими мутациями накапливается сукцинат, что может привести к формированию нейроэндокринных опухолей таких, как параганглиома, карцинома почек и стромальная опухоль желудочно-кишечного тракта.
Предложены последовательности области гена SDHA из клеток параганглиомы, полученной от пяти особей лебедя-шипуна (Cygnus olor). Последовательности получали методом ПЦР с праймеров и секвенировали методом Сэнгера. Проведите множественное выравнивание последовательностей совместно с последовательностью дикого типа (NC_049170.1 complement(92788988..92803671)) через модуль MUSCLE, находящийся в UGENE.
Задача. Определите наиболее часто встречающуюся мутацию, которая может приводить к образованию параганглиомы у лебедя-шипуна.
Ответ введите в формате c.XY>Z, где X — позиция нуклеотида, Y — истинный нуклеотид, Z — мутантный нуклеотид.
Последовательности (\(5^\prime\rightarrow3^\prime\)) мутантных вариантов гена SDHA из Cygnus olor:
- SDHA_mut1 CCTGATGCTTTGTGCTCTACAAACAATCTATGGTGCTGAGGCTCGCAAGGAGTCCCGGGGTGCTTATGCCAGAGAGGACTATAAGGTACGGA.
- SDHA_mut2 CCTGATGCTTTGTGCTCTACAAACAATCTATGGTGCTGAGGCTCGCAAGCAGTCCCGGGGTGCTTATGCCAGAGAGGACTATAAGGTACGGA.
- SDHA_mut3 CCTGATGCTTTGTGCTCTACATACAATCTATGGTGCTGAGGCTCGCAAGGAGTCCCGGGGTGCCTATGCCAGAGAGGACTATAAGGTACGGA.
- SDHA_mut4 CCTGATGCTTTGTGCTCTACATACAATCTATGGTGCTGAGGCTCGCAAGGAGTCCCGGGGTGCTTATGCCAGAGAGGACTATAAGGTACGGA.
- SDHA_mut5 CCTGATGCTTTGTGCTCTACAAACAATCTATGGTGCTGAGGCTCGCAAGGAGTCCCGTGG TGCTTATGCCAGAGAGGACTATAAGGTACGGA.
Список литературы
- Скачивание последовательности дикого типа: перейти по ссылке https://www.ncbi.nlm.nih.gov/nuccore/NC_049170.1?from=92788988&to=92803671&report=genbank&strand=true \(\Rightarrow\) send to \(\Rightarrow\) coding sequence \(\Rightarrow\) create file.
- Предлагаемый софт для решения задачи: UGENE http://ugene.net/ru/.
- MUSCLE: подаваемый файл должен содержать все последовательности (дикий тип и мутантные варианты) и иметь формат fasta.
c.1765T>C.
Трансформация бактерий — это процесс, при котором бактерия поглощает чужеродную ДНК из окружающей среды. Встраивание такой ДНК в генетический аппарат клетки позволяет бактериям приобретать новые свойства, такие как устойчивость к антибиотикам или дополнительные метаболические возможности.
В лабораторных условиях искусственная трансформация дает возможность вводить новые гены, изучать функции генома и создавать бактерии с полезными свойствами для биотехнологий. Существует два основных метода искусственной трансформации:
- Первый метод — химическая трансформация с тепловым шоком, когда с помощью химических веществ и теплового воздействия чужеродную ДНК вводят в клетку.
- Второй метод — электропорация, при которой электрический импульс временно делает мембрану клетки проницаемой, позволяя ДНК проникать внутрь.
Эффективность трансформации (ЭТ) оценивают по числу трансформированных клеток, выраженному в колониеобразующих единицах (КОЕ) на микрограмм (мкг) введенной ДНК (например, плазмидной).
\[\text{ЭТ} = \frac{\text{КОЕ}}{\text{количество~трансформируемой~плазмиды~в~мкг}}.\]
После приготовления химически и электрокомпетентных клеток E. coli штамма BL21 (DE3) проверяли эффективность их трансформации при помощи введения стандартного плазмидного вектора pUC19 (2686 пн). Для этого приготовили два разведения плазмиды из стоковой (исходной) концентрацией 508 нг/мкл: (1) развели в 1000 раз и (2) развели в 50 раз.
К электрокомпетентным клеткам добавили 2 мкл разведения плазмиды (1), а для химической трансформации использовали 5 мкл разведения плазмиды (2). После трансформаций клетки посеяли на чашки Петри следующим образом: после электропорации 1/20 от общего количества клеток, а химически трансформированных клеток — 1/10. После инкубации чашек Петри в термостате при 37 °С в течение 16 ч выросло 742 КОЕ после электропорации и 984 КОЕ после химической трансформации.
Задача. Определите, во сколько раз отличается эффективность трансформации методом электропорации от химического метода. Ответ округлите до целого числа.
75.
Умение анализировать научную информацию, работать с литературными источниками, письменно излагать свои мысли — является крайне востребованным навыком для молодого исследователя. Опыт проведения отборочного тура НТО показал, что написание короткого текста с развернутым ответом на вопрос оказалось одним из самых трудных и интересных заданий.
Нобелевская премия по химии в 2024 году присуждена за работы, связанные с компьютерными технологиями предсказания и укладки (фолдинга) белка.
Задание. Напишите текст (эссе) объемом не более 3000 символов (с учетом пробелов) на тему: «В чем отличия инструментов AlphaFold и Rosetta, за разработку которых была присуждена Нобелевская премия? В чем преимущества AlphaFold?»
Проведите анализ литературных данных. Сопроводите текст ссылками на авторитетные источники. Аргументируйте точку зрения.
- Длина эссе — не более 3000 символов (с учетом пробелов).
- Правильная аргументация, обоснование ответа.
- Отсутствие биологических ошибок.
- Ссылки на литературные источники.
- Текст написан лично автором, отсутствуют заимствованные фрагменты текста.
- Эссе, имеющие явные признаки использования ChatGPT, Алиса ГПТ и подобных инструментов, могут быть оценены в 0 (ноль) баллов.
- В зачет идет одно лучшее эссе от команды.
- При наличии нескольких одинаковых/стереотипных текстов, будет оцениваться текст, загруженный первым, остальные будут оценены в 0 баллов.
- При совпадении фрагментов текстов у разных команд будет оценен первый загруженный текст, остальные будут оценены в 0 баллов.
- При превышении рекомендованной длины текста оцениваться будут первые 3000 символов (с учетом пробелов).
- Список литературы включается (!) состав в 3000 символов.
Командные задачи третьего блока для 10–11 классов открыты для решения. Соревнование доступно на платформе Яндекс.Контест: https://contest.yandex.ru/contest/70129/enter/.
Лактаза — это фермент, гидролизующий гликозидные связи между остатками галактозы и глюкозы в молекулы лактозы — основного молочного сахара. Недостаток этого фермента приводит к непереносимости лактозы.
У человека фермент лактаза транслируется в виде пре-фермента, содержащего сигнальный пептид для эффективного транспорта белкового продукта. После доставки сигнальный пептид отщепляется.
Задача. Рассчитайте изоэлектрическую точку сигнального пептида лактазы. Для выполнения задания используйте последовательность мРНК лактазы и вспомогательную информацию из базы данных NCBI (NM_002299.4). Ответ округлите до целого значения.
P. S. Изоэлектрическая точка — величина pH среды, при которой суммарный электрический заряд пептида равен нулю. \[pI=\frac{\sum pKa_i}{n}= \frac{pKa_1+pKa_2+pKa_3+\cdots}{n} ,\] где \(pKa\) — константа диссоциации для полярных групп в пептиде, \(n\) — количество полярных групп в пептиде.
Справочная таблица со значениями \(pKa\) для аминокислот: https://disk.yandex.ru/i/ZUYKDsDFQDJmRA.
6.
Филогенетическое дерево — это графическое представление эволюционных связей между видами, популяциями или генами, основанное на сходстве их биологических характеристик, таких как последовательности ДНК или белков. Эти деревья помогают понять, как виды эволюционировали от общего предка, проследить генетическое разнообразие, а также исследовать процессы, связанные с изменениями в геномах. Одним из первых шагов в построении филогенетического дерева является вычисление матрицы расстояний, где каждое значение отражает степень различий между последовательностями. Например, расстояние Хэмминга измеряет количество позиций, в которых символы двух последовательностей различаются. Эта матрица затем используется для построения самого дерева.
Метод UPGMA (Unweighted Pair Group Method with Arithmetic Mean) объединяет виды или группы с минимальным расстоянием и создает новое значение для кластеров, основываясь на среднем арифметическом. Повторяя этот процесс, UPGMA постепенно объединяет все виды в дерево, отображающее их эволюционные связи. Таким образом, построение матрицы расстояний и использование метода UPGMA — это два последовательных этапа, необходимых для анализа эволюционной истории на основе молекулярных данных. В этой задаче предстоит построить матрицу дистанций между последовательностями, а в следующей задаче — выполнить построение филогенетического дерева методом UPGMA.
Задача. Напишите программу, которая принимает на вход последовательности ДНК, попарно сравнивает все последовательности и определяет количество различий между ними (расстояние Хэмминга). Результаты попарного сравнения программа должна записать в симметричную матрицу \(N \times N\) (где \(N\) — количество последовательностей). Каждый элемент этой матрицы \(D_{ij}\) — это количество различий между \(i\)-й и \(j\)-й последовательностями.
Формат входных данных
Программа должна принять (через input()) строку, содержащую произвольное количество последовательностей ДНК (больше двух), разделенных пробелами.
Формат выходных данных
Программа должна вывести (через print()) матрицу дистанций в виде списка списков.
Тестовые данные
| Номер теста | Стандартный ввод | Стандартный вывод |
|---|---|---|
1 |
AGCT ACCT AGTT TGCA |
[[0, 1, 1, 2], [1, 0, 2, 3], [1, 2, 0, 3], [2, 3, 3, 0]] |
Ниже представлено решение на языке Python.
def hamming_distance(seq1, seq2):
"""Calculates the Hamming distance between two sequences."""
return sum(el1 != el2 for el1, el2 in zip(seq1, seq2))
def compute_distance_matrix(dna_sequences):
"""Creates a symmetric distance matrix for DNA sequences."""
n = len(dna_sequences)
matrix = [[0] * n for _ in range(n)]
for i in range(n):
for j in range(i + 1, n):
distance = hamming_distance(dna_sequences[i], dna_sequences[j])
matrix[i][j] = distance
matrix[j][i] = distance # Symmetric value
return matrix
# Input DNA sequences
input_data = input()
dna_sequences = input_data.split()
# Calculate and print the distance matrix
result_matrix = compute_distance_matrix(dna_sequences)
print(result_matrix)
Напишите программу, которая принимает на вход одну строку, содержащую последовательности ДНК, разделенные пробелами. Необходимо рассчитать матрицу расстояний между этими последовательностями (где расстояние определяется как количество различий — расстояние Хэмминга), а затем использовать эту матрицу для построения филогенетического дерева методом UPGMA (Unweighted Pair Group Method with Arithmetic Mean). На выходе программа должна вывести финальное филогенетическое дерево в формате строки, где узлы дерева представлены в скобках, а листья — номерами последовательностей (их позиции в исходной строке, начиная с 1). Дерево должно быть строго бинарным, а порядок объединения пар в скобках определен по возрастанию номеров.
Формат входных данных
Программа должна принять (через input()) строку, содержащую произвольное количество последовательностей ДНК (больше двух), разделенных пробелами.
Формат выходных данных
Программа должна вывести (через print()) полученное филогенетическое дерево.
Тестовые данные
| Номер теста | Стандартный ввод | Стандартный вывод |
|---|---|---|
1 |
AGCT ACCT AGTT TGCA |
(((1,2),3),4) |
Ниже представлено решение на языке Python.
def cd(str1, str2):
return sum(el1 != el2 for el1, el2 in zip(str1, str2)) + abs(len(str1) - len(str2))
def nop(seq):
s = len(seq)
t = [[0] * s for _ in range(s)]
for i in range(s):
for j in range(i + 1, s):
d = cd(seq[i], seq[j])
t[i][j] = d
t[j][i] = d
return t
def minc(t):
m = float("inf")
x, y = -1, -1
for i in range(len(t)):
for j in range(len(t[i])):
if t[i][j] < m and i != j:
m = t[i][j]
x, y = i, j
return x, y
def jl(l, a, b):
if b < a:
a, b = b, a
l[a] = "(" + l[a] + "," + l[b] + ")"
del l[b]
def jt(t, a, b):
if b < a:
a, b = b, a
row = []
for i in range(0, a):
row.append((t[a][i] + t[b][i]) / 2)
t[a] = row
for i in range(a + 1, b):
t[i][a] = (t[i][a] + t[b][i]) / 2
for i in range(b + 1, len(t)):
t[i][a] = (t[i][a] + t[i][b]) / 2
del t[i][b]
del t[b]
def upgma(t, l):
while len(l) > 1:
x, y = minc(t)
jt(t, x, y)
jl(l, x, y)
return l[0]
inp = input()
seq = inp.split()
dt = nop(seq)
l = [str(i+1) for i in range(len(seq))]
otv = upgma(dt, l)
print(otv)
При обработке больших неидентифицированных ранее молекул ДНК, например, геномов новооткрытых бактерий или фагов, бывает полезно примерно оценить вероятность встречи того или иного сайта рестрикции. Это позволяет правильно подобрать рестриктазы для построения карты генома.
Определите, во сколько раз отличается количество вероятных сайтов рестрикции MslI в геноме E. coli от количества вероятных сайтов этой же рестриктазы в геноме фага-лямбда, который заражает данную бактерию.
Предположим, что нуклеотиды в геномах расположены случайно и количество A = T = C = G. Таким образом, важны только длины геномов. В ответе укажите целое число, при необходимости округлите результат до ближайшего целого числа.
Список литературы
- Геном E. coli — https://www.ncbi.nlm.nih.gov/nuccore/U00096.2 (pdf-версия https://disk.yandex.ru/d/vr8qwlPDHTjJJA).
- Геном фага лямбда — https://www.ncbi.nlm.nih.gov/nuccore/J02459 (pdf-версия https://disk.yandex.ru/d/t8Tpr1_RPyvjjw).
96.
В смесь для ПЦР добавляют dUTP вместо (или вместе с) dTTP для того, чтобы предотвращать перекрестную контаминацию «чужими» образцами. Проанализируем, что произойдет, если добавить реакционную смесь для ПЦР dTTP и dUTP, а потом очистить продукты реакции от «контаминированных».
Для реакции ПЦР использовали мастер-микс 5х, содержащий Taq ДНК-полимеразу с «горячим стартом», смесь dNTP (dATP, dGTP и dCTP — 0,6 мM, dTTP/dUTP — 0,3 мM), 15 мМ ионы \(\ce{Mg^{2+}}\), реакционный буфер, глицерин, красители. В реакционную смесь объемом 25 мкл, содержащую 1х мастер-микс, добавили прямые (ATG) и обратные (GCA) праймеры по 0,4 мкМ и ДНК-матрицу: ATGACGAGGTCCTGC — 5 копий на 1 мкл реакционной смеси. После 10 раундов репликации полученную смесь обработали UDG — урацил-ДНК-гликозилазой, которая гидролизует урацил в составе ДНК.

Задача. Определите процент продуктов, подвергшихся гидролизу UDG, если ДНК-полимераза включает dUTP каждый третий комплементарный нуклеотид от начала синтеза новой цепи. В ответе приведите целое число. Пример ответа: 100.
100.
В результате воздействия различных факторов, например, ультрафиолета или активных форм кислорода, ДНК в клетке нарушает свою структуру. Наиболее часто из-за этого образуются окислительные повреждения ДНК, в частности — 8-оксогуанин.
Количество окислительных повреждений в одной клетке может достигать 10\(^6\)–10\(^7\) повреждений в сутки. Чтобы исправить такое огромное количество повреждений, внутри клетки работают ферменты эксцизионной репарации оснований (base excision repair — BER). За удаление 8-оксогуанина в человеческих клетках отвечает 8-оксогуанин-ДНК-гликозилаза (hOGG1, h — human).
Как известно, для многих ферментов существуют гомологи из других организмов, что характерно и для hOGG1. В клетках E. coli также присутствует система BER, а ДНК-гликозилаза Fpg (DNA-formamidopyrimedine-glycosylase — формамидо-пиримидин-ДНК-гликозилаза) имеет схожую каталитическую активность c hOGG1. В отличие от других гомологичных белков, hOGG1 и Fpg имеют совершенно разные аминокислотные последовательности, а также вторичную и третичную структуру.
Таким образом, данные ферменты гомологичны по функциям, но различаются. Чтобы частично продемонстрировать это, проведем следующий эксперимент.
Для сравнения первичной структуры ферментов OGG1 и Fpg их независимо обработали химотрипсином и проанализировали продукты реакции методом белкового гель-электрофореза. Среди множества продуктов каждой реакции определите их моду и в ответе укажите абсолютную разницу (модуль) между данными показателями.
Взяв последовательности полипептидов, необходимо расщепить их в соответствии с указанными условиями. Для каждого пептида выбрать наиболее часто встречающуюся длину продуктов гидролиза и оценить разность.
Список литературы
- human OGG1 — https://www.ncbi.nlm.nih.gov/nuccore/U88527 (pdf-версия https://disk.yandex.ru/i/jfpoybkzjyXKxQ).
- Fpg E. coli — https://www.ncbi.nlm.nih.gov/nuccore/NC_000913.3?from=3810343&to=3811152&report=genbank&strand=true (pdf-версия https://disk.yandex.ru/i/Hi0llFltgdWn9Q).
- Статья в Википедии про моду — https://ru.wikipedia.org/wiki/Мода_(статистика).
3.
В биохимии важно измерение кинетических параметров ферментативных реакций.
К одной из наиболее известных и простых моделей ферментативной кинетики относят модель Михаэлиса – Ментен, предложенную в 1913 году. Она объясняет характерную гиперболическую зависимость активности фермента от концентрации субстрата и позволяет получать константы, которые количественно характеризуют эффективность фермента. Простейшая кинетическая схема, для которой справедливо уравнение Михаэлиса – Ментен: \[E + S \Leftrightarrow ES \longrightarrow E + P,\] где \(E\) — фермент, \(S\) — субстрат, \(ES\) — комплекс фермент-субстрат, \(P\) — продукт.
Кинетическое уравнение имеет следующий вид: \[v=\frac{V_m \cdot S}{S + K_m},\] где \(v\) — скорость реакции, \(V_m\) — максимальная скорость реакции, \(K_m\) — константа Михаэлиса (численно равна концентрации субстрата, при которой скорость реакции составляет половину от максимальной), \(S\) — концентрация субстрата.
Константа \(K_{m}\) показывает сродство фермента к субстрату, т. е. насколько хорошо фермент связывает субстрат. \[K_m=\frac{k_{-1}+k_2}{k_1},\] где \(k_{-1}\) — константа скорости реакции распада фермент-субстратного комплекса на фермент и исходный субстрат, \(k_{1}\) — константа скорости реакции образования фермент-субстратного комплекса, \(k_2\) — константа скорости реакции распада фермент-субстратного комплекса на фермент и продукт. Далее будем называть \(k_{2}\) — \(k_{cat}\) (каталитическая константа).
Как известно, для ДНК-полимераз субстратами выступают ДНК-матрица и dNTP, и принято измерять точность \(k_{ins}\) (ins — точность включения — insertion), которая состоит из показателей специфичности включения неверного нуклеотида \(k_{sp,w}\) (w — wrong) к верному \(k_{sp,r}\) (r — right).
Специфичность: \[k_{sp}=\frac{k_{cat}}{K_m}.\]
Точность: \[k_{ins}=\frac{k_{sp,w}}{k_{sp,r}}.\]
Для ферментативной реакции включения dCTP и dATP напротив 8-оксогуанина на субстрате типа «праймер-матрица» фрагментом Кленова ДНК-полимеразы I E. coli измерили кинетические параметры в условиях кинетики Михаэлиса – Ментен. Результаты представлены в таблице 1.1.
| dNTP | [S], nM | \(V \times 10^{-4}\), nM/sec | \(V_{\text{max}}\), nM/sec | \(K_{\text{cat}}\), s\(^{-1}\) \(\times 10^{-3}\) |
|---|---|---|---|---|
| dATP | 100 | 58,33 | 0,056 | 282 |
| dCTP | 100 | 4,31 | 0,004 | 479 |
Определите точность включения dATP напротив 8-оксогуанина фрагментом Кленова ДНК-полимеразы I E. coli на субстрате «праймер-матрица». В ответе укажите число с точностью до двух знаков после запятой (десятичный разделитель — точка).
Список литературы
- Статья про уравнение Михаэлиса – Ментен: https://ru.wikipedia.org/wiki/Уравнение_Михаэлиса_—_Ментен.
0.57.
Выберите верные утверждения о подборе праймеров для получения ПЦР-продукта с геномной ДНК и дальнейшего клонирования в плазмидный вектор для последующей продукции белка:
- Праймеры могут отжигаться в любом месте, главное, чтобы последовательность гена находилась между ними.
- Праймеры должны иметь температуры плавления, различающиеся не более, чем на 5 °C.
- Праймеры должны обязательно иметь одинаковую длину — не более 20 нт.
- GC-состав прямого и обратного праймера может отличаться, главное, чтобы он был менее 30%.
- Температура отжига праймера вычисляется по формуле \(\text{T}=2(\text{A}\,+\,\text{T})\,+\,4(\text{G}\,+\,\text{C})\).
- Праймеры должны отжигаться ровно на начало и конец гена.
B, F.
Основываясь на предыдущем задании, подберите наилучшие праймеры для получения ампликона гена BGAL11, кодирующего бета-галактозидазу в Arabidopsis thaliana, для клонирования в плазмидный вектор и последующей наработки белка.
Задача. Выберите наилучшую пару праймеров, в которую добавлены оптимальные сайты рестрикции.
Последовательности праймеров даны в направлении \(5^\prime\rightarrow3^\prime\).
Комментарий. Порядок приоритета параметров для подбора праймеров от наиболее значимого до менее значимого — Tm, длина, GC-состав.
Выберите верные варианты ответа:
- GGTCGTGGGAAGAAGAACTGA.
- GGCTTTTAACAGCTGTTC.
- TTTTCACAGAGACATTGCC.
- ATGAGAAAACATTCTCTTGATCG.
- CAAGTGTGGTCGTGGGAAG.
- ATGAGAAAACATGGAGGATCTGC.
- GCTTTATCCCGTGCACCTTAAAC.
- TGAGAAAACATTCTCTTGAT.
- TCAGTTCTTCTTCGCTTACGG.
- GGCAATGTCTCTGTGAAAA.
- CGATCAAGAGAATGTTTTCTCAT.
- TGGATGGTACACAACA.
- TCAGTTCTTCTTCCCACGACC.
- GTAAACTCGAAGGTA.
- ATGAGAAAACATTCTCTT.
Список литературы
- Ссылка на последовательность гена BGAL11 — https://www.ncbi.nlm.nih.gov/nuccore/NC_003075.7?from=16667837&to=16672188&report=genbank&strand=true (pdf-версия https://disk.yandex.ru/i/bcQimJA0H0dVJQ).
- Возможный инструмент для решения задачи: UGENE http://ugene.net/ru/.
4,13.
Во второй части данного задания были подобраны праймеры для получения ампликона гена BGAL11, но при реакции ПЦР с такими праймерами будут получены продукты с тупыми концами. Этот продукт невозможно клонировать вставку в плазмиду, поскольку могут возникнуть такие проблемы, как неверное направление вставки относительно промотора, самолигирование вектора/вставки из-за большой длины линейных фрагментов, включение нескольких копий вставки в вектор. Кроме этого, клонирование по тупым концам имеет намного более низкую эффективность, чем по липким концам. Для решения данных проблем генные инженеры добавляют к праймерам сайты рестрикции для того, чтобы получить липкие концы после проведения ПЦР.
В лаборатории имеется библиотека доступных рестриктаз, список которых представлен ниже. Необходимо выбрать наилучшие сайты рестрикции для клонирования гена BGALL11 в плазмидный вектор pBlueScript SK(–) с геном STAT2 (позиция в плазмиде 218–2749 пн) [1].
Задача. Вырежите ген STAT2 и вставьте вместо него ген BGAL11 под промотор T3.
Библиотека лабораторных рестриктаз:
- AfeI.
- BamHI.
- ClaI.
- DraIII.
- EcoRI.
- EcoRV.
- HaeI.
- HauII.
- HindIII.
- KpnI.
- MfeI.
- NcoI.
- NdeI.
- SalI.
- XbaI.
В ответе укажите номер рестриктазы с обозначением прямого (F) и обратного (R) праймеров, разделенных дефисом. Пример ответа: 1F-2R.
Список литературы
- Карта плазмиды pBlueScript SK(–) STAT2 — https://www.addgene.org/8705/ — Addgene #8705 (или https://disk.yandex.ru/i/UaMSyG7d919CWw).
15F-14R.
В 2020 году Нобелевская премия по химии была присуждена Дженнифер Дудне и Эммануэль Шарпаньте за открытие системы CRISPR-Cas9 у бактерии Streptococcus pyogenes. Это открытие дало генным инженерам и молекулярным биологам удобный инструмент для направленного редактирования генома организмов.
В 2024 году Нобелевскую премию по химии дали Дэвиду Бэйкеру, Дэмису Хассабису и Джону Джамперу. Дэмис Хассабис и Джон Джампер являются основателями компании DeeepMind и получили премию за разработку нейросетевых алгоритмов для предсказания структуры белков по их первичной аминокислотной последовательности. Созданная ими в 2020 году нейронная сеть AlphaFold2 произвела прорыв в предсказании структуры белка по последовательности аминокислот. В мае 2024 года DeepMind анонсировала новую нейронную сеть AlphaFold3, которая может предсказывать структуру не только одиночных белков, но и белок-белковых, белок-нуклеиновых комплексов и даже комплексов белок-лиганд для некоторых молекул. Компания предоставила октрытый доступ к серверу AlphaFold Server (https://alphafoldserver.com/) для ученых со всего мира. Каждому Google-аккаунту доступно 20 моделирований в день.
Необходимо воспользоваться онлайн-сервисом AlphaFold3 Server, чтобы предсказать структуру комплекса белка SRISPR-Cas9 из Streptococcus pyogenes с гидовой РНК (gRNA) и целевой последовательностью двуцепочечной ДНК, а затем ответить на несколько вопросов.
Последовательность белка SRISPR-Cas9 из Streptococcus pyogenes:
Последовательность гидовой РНК: GGCGCAUAAAGAUGAGACGCGUUUUAGAGCUAUGCUGUUUUGAAAAAAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUCG.
Последовательности двуцепочечной ДНК мишени (обе цепи записаны в направлении \(5^\prime\rightarrow 3^\prime\)): GGCGCATAAAGATGAGACGCTGGCGATTAG CTAATCGCCAGCGTCTCATCTTTATGCGCC.
Задача. Произведите структурное выравнивание и определите показатель RMSD расчетных моделей с экспериментальной. PyMOL выдает все линейные размеры в Ангстремах — параметр RMSD не исключение. Посчитайте среднее отклонение для пяти моделей, ответ приведите в пикометрах и округлите до целых. В ответе укажите только число.
Любое значение в промежутке 215–235.





